 2016, Vol. 51
2016, Vol. 51

20世纪60年代美国发动侵略越南战争,当地疟疾肆虐,疟原虫对已有药物产生耐药,使战斗力严重减弱。应越南要求中国提供有效抗疟药物,我国政府决定全国范围研究新型抗疟药,遂于1967年5月23日成立了研究协作组,简称“523任务”,涵盖60多个研究单位,500多位研究人员。在由启动研究到临床实验和应用的整个研发过程,统一由“523任务”调度,并非固定在一个研究单位中 (张文虎. 创新中的社会关系: 围绕青蒿素的几个争论. 自然辩证法通讯. 2009,31: 32-39)。
1.2 从中药和民间药寻找药物或先导物在“523任务”的诸多研究项目中,有一个课题是“民间防治疟疾有效药物疗法的重点调查研究”,这个研究小组获得了许多苗头,例如从植物鹰爪分离出有效抗疟单体鹰爪甲素,从陵水暗罗中分离出暗罗素的金属化合物,对常山乙碱的结构改造,以及青蒿素等。
1.3 青蒿和青蒿素的发现1969年中国中医研究院中药研究所加入“中医中药专业组”。组长屠呦呦和组员余亚纲等从中医药古籍中搜集并筛选中草药单、复方数百种中,余亚刚和顾国明发现青蒿呈现高频率 (唐宋元明的医籍、本草和民间都曾提到有治疟作用)。通过广泛实验筛选,聚焦到青蒿的乙醇提取物,对疟原虫抑制率达到60%~80%,虽然活性重复性差,但为后来研究提供了有价值的参考 (李国桥等. 青蒿素类抗疟药. 北京: 科学出版社,2015: 3)。
屠呦呦从东晋葛洪《肘后备急方》阐述青蒿的用法得到了启发,“青蒿一握,以水二升渍,绞取汁,尽服之”,冷榨服用“绞汁”,悟出可能不宜高温加热的道理,并考虑到有效成分可能在亲脂部分,遂改用乙醚提取,于1971年10月在去除了酸性成分的中性提取物中,分离得到的白色固体对鼠疟原虫的抑制率达100%。由绞汁联想低温提取,但由水浸的冷榨液 (通常含有水溶性成分) 怎样推论是脂溶性成分,文献中无从考证。不过选择乙醚为萃取剂,无疑是发现青蒿素、开辟青蒿素类药物治疗的关键一步。
用乙醚从黄花蒿分离出的倍半萜化合物除青蒿素 (1,artemisinin) 外,还鉴定了其他成分,有青蒿酸 (2,arteannuic acid)、青蒿甲素 (3,arteannuin A)、青蒿乙素 (4,arteannuin B)、青蒿丙素 (5,arteannuin C) 和紫穗槐烯 (6,amorphane) 等。这些倍半萜除2是酸性化合物外,都是中性成分,色谱柱分离单体后确证了结构,但没有或只有很弱的抗疟活性 (屠呦呦,倪慕云,钟裕容等. 中药青蒿化学成分的研究. 药学学报,1981,16: 366-370)。

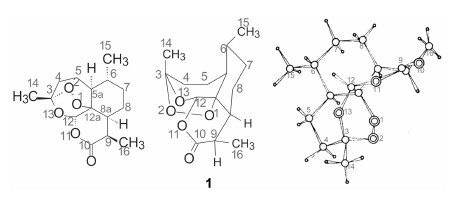
青蒿素为白色针状结晶,熔点151~153 ℃,元素分析和质谱表明分子式为C15H22O5,不溶于水,溶于丙酮、乙醇、乙醚、石油醚和碱性水溶液中,NaOH溶液滴定可消耗1摩尔当量。定性分析可氧化FeCl2或NaI呈现颜色反应,与三苯膦定量测定生成等摩尔量的氧化产物,提示青蒿素含有氧化性功能基。
2.2 谱学行为紫外光谱未显示有芳环的共轭体系,红外光谱显示有δ内酯型的羰基峰,13C核磁共振谱表明有15个C原子信号,伯仲叔季碳分别为3、4、5和3个。其中一个季碳以羰基存在,另两个峰在低场79.5和105 ppm,提示氧原子连于其上。在高场处的5个叔碳为二重峰。1H核磁谱在5.68 ppm处有单峰,表明具有-O-CH-O-片段。从倍半萜的生源推测含有共用4个氧原子的缩酮、缩醛和内酯结构,但难以归属第五个氧原子。
彼时 (1975年) 中国医学科学院药物研究所的523组于德泉报告了另一抗疟活性成分鹰爪甲素的结构,是含有过氧键的化学成分 (梁晓天,于德泉,吴伟良等. 鹰爪甲素的化学结构. 化学学报,1979,37: 215-230),这对测定青蒿素结构以巨大启示,结合定性分析的氧化性,联想青蒿素结构中也含有一个过氧键。推测有以下3种可能 (1,7,8)。

确定该过氧键所处的位置,是中国科学院生物物理研究所的523组经X-射线晶体衍射分析,进而经旋光色散 (ORD) 分析,最终确定了青蒿素的化学结构和绝对构型 (中国科学院生物物理所抗疟药青蒿素协作组. 青蒿素晶体结构及其绝对构型. 中国科学,1979,(11): 1114-1128; 刘静明,倪慕云,樊菊芬等. 青蒿素 (arteannuin) 的结构和反应. 化学学报,1979,37: 129-142; 青蒿素结构研究协作组. 一种新型的倍半萜内酯——青蒿素. 科学通报,1977,22: 142)。
青蒿素在Pd/CaCO3催化下氢化,将过氧键还原成醚键,生成的产物命名为还原青蒿素,推测反应机制如图 1,还原青蒿素的结构与天然存在的青蒿丙素 (5) 结构相同。
 | Figure 1 还原青蒿素的生成机制 |
低温下青蒿素与NaBH4反应,将C10的羰基还原成以半缩醛形式存在的羟基化合物 (9),称作二氢青蒿素。但在Lewis酸存在下,用NaBH4处理,C10羰基还原成亚甲基化合物 (10); 在乙酸-硫酸作用下,发生失碳和重排,生成化合物11。生成11的反应历程如图 2所示。
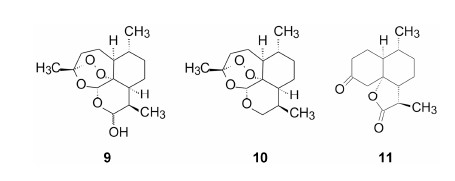
 | Figure 2 酸催化青蒿素失碳重排反应 |
我国首先实现青蒿素全合成的是上海有机化学研究所许杏祥等由青蒿酸 (2) 为起始物完成的。基于生源原理,2应是青蒿素的生物合成前体,黄花蒿中2的含量较高也是个佐证。图 3是合成路线的简要过程。青蒿酸2经酯化得2a,硼氢化钠将环外双键还原成2b,臭氧氧化开环,得到单环的醛酮物2c,酮基选择性用丙二硫醇保护,得2d,2d的醛基用原甲酸三甲酯处理得到烯醇醚2e,去除硫醚保护基的2f,经O2光氧化得到关键中间体缩醛过氧化物2g,酸水解2g,发生级联的关环反应,形成过氧桥环、环醚和内酯环,生成青蒿素1 (Xu XX,Zhu J,Huang DZ,et al. Studies on structure and syntheses of artennuin and related compound. 10. The stereocotrolled synthesis of artennuin and deoxy artennuin from arteneuic acid. Acta Chim Sin (化学学报),1983,41: 574-576)。与此同时,Schmid和Hofheinz以不同的合成方法也完成了青蒿素的全合成 (Schmid G,Holheinz W. Total synthesis of Quinghaosu. J Am Chem Soc,1983,105: 624-625)。以后又陆续报道了不同的合成路线和改进方法。青蒿素全合成的成功,是对化学结构的最终确证,也为实现工业化生产开辟了道路。
 | Figure 3 由青蒿酸为原料合成青蒿素的路线 |
屠呦呦研究组用乙醚提取的中性成分进行了动物安全性实验和健康志愿者试服后,在海南和北京进行了30例患者的治疗,虽然治愈率不高,但证实有明确的抗疟作用。中性成分中含有屠呦呦研究组后来提纯的青蒿素 (当时称作青蒿素II)。这意味着从物质基础到临床作用开创了青蒿素治疗疟疾的道路。
3.2 青蒿素的规模制备和临床疗效的确证在醚提取物和初步疗效的启示下,云南药物研究所罗泽渊等和山东中医药研究所魏振兴等用汽油或乙醚提取当地的黄花蒿,得到了纯度高的青蒿素,显著提高了抗疟效价。特别是罗泽渊等发明的“溶剂汽油法”,为提供临床研究和大规模生产奠定了技术和物质基础。
广州中医学院“523组”李国桥等在云南开展脑型疟防治研究,与提供临床用药的罗泽渊合作,进行了青蒿素的抗疟临床研究。他们给患者口服青蒿素 (原称黄蒿素),发现恶性疟原虫纤细环状体停止发育并迅速减少的现象,认定青蒿素对恶性疟原虫的速杀作用远超奎宁和氯喹。治疗收治的18例患者,其中有1例脑型恶性疟、2例黄疸型恶性疟和11例非重症恶性疟,4例间日疟,全部迅速临床治愈,标志着首先临床证实青蒿素治疗恶性疟的疗效及其速效、低毒的特点,是对青蒿素临床应用价值的重要发现 (张剑方. 迟到的报告——五二三项目与青蒿素研发纪实. 广州: 羊城晚报出版社,2006)。
4 以青蒿素为先导物的结构优化青蒿素为倍半萜,结构中5个氧原子交织形成环醚、过氧环醚、环状缩醛、环状缩酮和内酯。由于分子中缺乏助溶基团,因而水溶性低,而且脂溶性也不强。虽然中国药监部门于1985年批准为新药上市,但生物利用度和生物药剂学性质差,限制了青蒿素的临床应用。因而以青蒿素为先导物作结构优化是发展青蒿素类药物的必然趋势。研究青蒿素抗疟的作用机制由于相对滞后,结构优化是以表型变化和分析构效关系进行的。
4.1 过氧键的存在和特异的骨架支撑是重要的结构因素用化学方法确定青蒿素结构,将过氧键转变成醚键、生成的还原青蒿素 (5),经活性评价,完全失去了抗疟作用,参考有抗疟活性的鹰爪甲素也含有过氧键,推论过氧键应是必需的药效团; 不过当筛选更多的过氧化合物,并非含过氧结构的分子都有活性,提示支撑过氧结构的分子骨架也有特异性贡献。因而青蒿素的优化以保存过氧键和分子骨架为前提,设计合成其衍生物。
4.2 我国研制的青蒿素类药物: 蒿甲醚在证明结构的化学反应中,用NaBH4还原青蒿素的酯羰基,得到半缩醛化合物二氢青蒿素 (9),抗疟活性强于青蒿素一倍,提示变换内酯基团仍可保持并提高活性。为了提高化合物的抗疟活性和稳定性,李英等从化合物9出发合成二氢青蒿素的3类衍生物。
4.2.1 C10醚类化合物二氢青蒿素在BF3-乙醚催化下与醇反应生成缩醛醚(通式12),产物为一对差向异构体(α,β-异构体),是稳定的化合物,主产物为β构型。醚化合物分离成α和β单体对鼠疟 (Plasmodium. berghei) 抗氯喹原虫株作活性评价,以青蒿素为阳性对照,测定抑制90% 原虫的所需剂量 (SD90)。表 1列出了有代表性的C10含醚基的化合物的活性。
|
|
Table 1 C10醚类化合物的结构及其活性 |
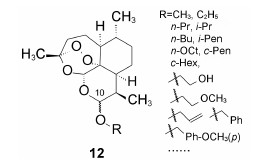
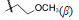
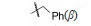
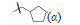
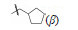

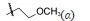
表 1的数据提示,二氢青蒿素 (9) 的活性大约强于青蒿素1倍,甲醚化合物 (13) 的抗疟活性强于二氢青蒿素2倍。醚基的R烷基增大,活性降低; 羟乙基醚化合物21没有活性。β差向体的活性一般略高于相应的α差向体。R为CH3的化合物13活性高于其他烷基取代,13称作蒿甲醚。
4.2.2 C10酯类化合物二氢青蒿素9在吡啶介质中与酸酐、酰氯或氯代甲酸酯反应得到C10酯类化合物 (通式22),生成的羧酸酯为单一的α差向体。与氯代甲酸酯反应得到C10碳酸酯也以α差向体为主,温度稍高,产生少量β体。
表 2列出了二氢青蒿素的单酯和碳酸酯化合物的活性。这些酯类化合物的活性一般都比较高,并由此得到了如下的活性次序: 碳酸酯 > 羧酸酯 > 醚化合物 > 二氢青蒿素 > 青蒿素 (李英,虞佩林,陈一心等. 青蒿素类似物的研究. I. 还原青蒿素醚类、羧酸酯类和碳酸酯类衍生物的合成. 药学学报,1981,16: 429-439; 顾浩明,吕宝芬,瞿志祥. 青蒿素衍生物对伯氏疟原虫抗氯喹株的抗疟活性. 中国药理学报,1980,1: 48-50)。
|
|
Table 2 二氢青蒿素的单酯和碳酸酯化合物的活性 |


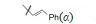
此外虞佩林等合成了9的取代苯甲酸酯化物,除化合物32的活性强于青蒿素10倍外,其余含硝基或卤素的衍生物活性一般 (虞佩林,陈一心,李英等. 青蒿素类似物的研究. IV. 含卤素、氮、硫等杂原子的青蒿素衍生物的合成. 药学学报,1985,20: 357-365; China Cooperative Research Group on Qinghaosu and Its Derivatives as Antimalarials. The chemistry and synthesis of Qinghaosu derivatives. J Tradit Chin Med (Eng Ed),1982,2: 9-16)。

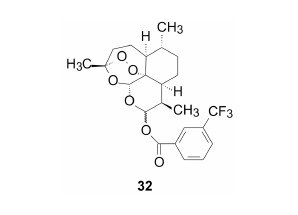
陈一心等用二元醇连接二氢青蒿素,设计合成了双二醚 (通式33),目标物的C10构型为α,β或β,β连接。活性评价表明均弱于单甲醚 (即蒿甲醚) [陈一心,虞佩林,李英等. 青蒿素类似物浓度研究. VII. 双 (二氢青蒿素) 醚和双 (二氢脱氧青蒿素) 醚类化合物的合成. 药学学报,1985,20: 270-273]。
4.2.4 蒿甲醚—候选化合物的确定上述二氢青蒿素的醚、羧酸酯和碳酸酯类衍生物中分别挑选出活性较高的13(β差向体)、24(α差向体) 和29(α差向体) 等3个化合物对伯氏疟原虫感染小鼠的治疗实验作进一步评价,表 3列出了这3个化合物与青蒿素和二氢青蒿素的活性数据。
|
|
Table 3 优选的化合物的抗疟作用比较。a: 抑制50% 疟原虫所需的剂量; b: 抑制90% 疟原虫所需的剂量; c: 小鼠每日给药, 5天后50% 疟原虫转阴的最低剂量; d: 小鼠每日给药, 5天后全部疟原虫转阴的最低剂量 |
表 3提示这3个化合物对感染小鼠都有良好的 治疗作用。进而评价对疟原虫感染食蟹猴的疗效,和对小鼠、大鼠、家兔、犬和猴的安全性试验,结合化合物的物化性质,确定蒿甲醚 (artemether) 为候选化合物,进入系统的临床前研究 (顾浩明,刘明章,吕宝芬等. 蒿甲醚在动物的抗疟作用和毒性. 中国药理学报,1981,2: 138-144)。1978年开始临床研究,并于1987年蒿甲醚以油针剂在我国批准为新药上市。
为了提高青蒿素的水溶性,便于注射用药,桂林制药厂刘旭以二氢青蒿素为原料合成了10多个C10羧酸酯,其中琥珀酸单酯 (代号804) 经红外、核磁、质谱和X-射线单晶衍射确定了结构,其钠盐可溶于水,适于注射给药。(刘旭. 青蒿素衍生物的研究. 药学通报,1980,15: 39; 濮金龙,陈荣光,蔡金玲. 青蒿素衍生物的结构测定. 广西药学会第二届年会学术讨论资料,P 82,1979年10月,转引自“青蒿琥酯的研究与开发. 刘旭主编”,桂林: 漓江出版社,2010: 52-53)。
临床前研究表明,青蒿琥酯 (34,artesunate) 有强效抗疟作用,对于抗氯喹的鼠疟有杀灭作用,青蒿琥酯钠对小鼠的LD50为1 003 mg·kg-1,家兔和犬的静脉注射亚急性毒性试验中,对体重、食欲、血象、肝肾功能和心脑电图未显示明显影响。其钠盐 (静脉滴注前用碳酸氢钠溶液溶解) 静脉注射后,在体内立即转化为二氢青蒿素。大鼠体内的血浆半衰期为15.6 min。犬的血浆半衰期为10~45 min。口服与静注青蒿琥酯后,消除半衰期分别为41.35和33.96 min。绝对生物利用度为40%。经临床研究,证明对间日疟、恶性疟和脑型疟均有效。1987年国家批准为新药上市 (杨启超,甘俊,李培青等. 青蒿素衍生物—青蒿酯的抗疟活性与毒性. 广西药学院学报,1981,(4): 1-6,转引自“青蒿琥酯的研究与开发. 刘旭主编”,桂林: 漓江出版社,2010: 56-63)。

蒿甲醚和青蒿琥酯虽然高效和速效抑制多种疟原虫感染,但在血浆中被迅速清除,半衰期短,以致患者体内的疟原虫不能完全清除而复燃。为此蒿甲醚、青蒿琥酯或二氢青蒿素常与其他长效的抗疟药合用,即所谓的基于青蒿素固定剂量的组合疗法 (ACT)。有代表性的固定配伍制剂有:
① 复方蒿甲醚片,是蒿甲醚 (13) 与本芴醇 (35,lumefatrine) 的固定制剂。本芴醇即是我国学者邓蓉仙等发明的抗疟药,有长效的特点 (邓蓉仙,余礼碧,张洪北等. 抗疟药的研究α-(烷氨基甲基)-卤代-4-芴甲醇类化合物的合成. 药学学报,1981,16: 920-924)。1984年由邓蓉仙、宁殿玺等研制组建的复方蒿甲醚片每片含蒿甲醚20 mg、本芴醇120 mg (李国桥等. 青蒿素类抗疟药.北京: 科学出版社,2015: 18)。经临床研究于1992年批准生产,英文商品名Coartem,每日服用1次,2002年WHO将Coartem列入第12版“基本药品目录”。2009年FDA批准在美国上市。
② 青蒿琥酯 (34) 与阿莫地喹 (36,amodiaquine) 的固定制剂,称作青蒿琥酯阿莫地喹片,每片含青蒿琥酯100 mg、阿莫地喹270 mg,商品名Coarsucum,每日两次,于2007年上市。
③ 二氢青蒿素 (9) 与哌喹 (37,piperaquine) 的固定制剂,含二氢青蒿素和哌喹分别为20 mg和160 mg,或40 mg和320 mg两种规格,商品名Eurartesim。
4.5 青蒿素类药物的作用机制青蒿素类药物的抗疟作用是通过影响疟原虫的膜系结构和线粒体,核膜和内网质出现肿胀和排列紊乱,阻断原虫摄取营养而导致氨基酸饥饿,同时迅速形成自噬泡并不断排出体外,原虫损失胞浆而死。从化学和生化机制分析,认为是通过血红蛋白的Fe2+介导,发生过氧键的裂解,产生自由基而起作用。原虫裂殖子进入红细胞,小滋养体的血红蛋白酶催化血红蛋白释放出血红素和Fe2+,青蒿素在Fe2+催化下过氧键裂解,生成氧和碳自由基,这些活性中间体抑制了消化液泡的生物膜和半胱氨酸蛋白酶。图 4是青蒿素 (及其相关药物) 在铁离子催化下经自由基两种途径的反应历程,生成的产物用方框标示 (Robert A,Dechy-Cabaret O,Cazelles J,et al. From mechanistic studies on artemisinin derivatives to new modular antimalarial drugs. Acc Chem Res,2002,35: 167-174)。
 | Figure 4 青蒿素 (类药物) 作用的生化机制 |
另有报道青蒿素的作用靶标是抑制了疟原虫钙ATP蛋白6 (PfATP6),PfATP6是SERCA类型的酶蛋白,它通过消耗ATP来调节疟原虫胞浆内钙离子浓度,保持钙水平的稳态。青蒿素类药物抑制PfATP6,从而引发疟原虫胞浆内钙离子浓度上升,起到杀灭疟原虫作用。研究表明疟原虫通过PfATP6基因突变而出现青蒿素耐药现象也为以上机制提供了佐证 (Dondorp AM,Yeung S,White L,et al. Artemisinin resistance: current ststus and scenarios for containment. Nat Rev Microbiol,2010,8: 272-280)。
5 其他青蒿素类抗疟药物的研究 5.1 蒿乙醚蒿乙醚 (14,artemotil,又称arteether),是二氢青蒿素C10-β-乙醚,由荷兰Brocacef公司研发,于2000年上市 (Brossi A,Venugopalan B,Dominguez Gerpe L,et al. Arteether a new anti-malarial drug: synthesis and anti-malarial properties. J Med Chem,1988,31: 645-650)。蒿乙醚以麻油制剂用于肌肉注射,治疗恶性疟原虫感染患者。3~12小时达到血浆峰浓度,血浆半衰期为1~2天,在肝脏经CYP3A4氧化脱乙基成二氢青蒿素。二氢青蒿素经葡醛酸苷化,经胆汁排出。作为跟进性药物的蒿乙醚,其疗效未显现优于蒿甲醚。
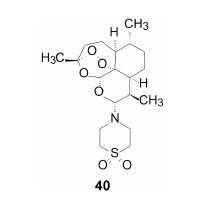
5.2 青蒿酮拜耳公司与香港科技大学合作研发青蒿素类药物,目标是改善既有药物的药代动力学。蒿甲醚、蒿乙醚和青蒿琥酯等药物在血浆中迅速水解,生成二氢青蒿素,后者经葡醛酸苷化而被清除,导致药效时间短。此外,二氢青蒿素还有一定的神经毒性。为了延长作用时间,减少二氢青蒿素的生成,合成了有代表性的化合物38~45,评价了抗疟活性,表 4列出了这些化合物的物化和抗疟活性 (Haynes RK,Fugmann B,Stetter J,et al. Artemisone-a high active antimalarial drug of the artemisinin class. Angew Chem Int Ed,2006,45: 2082-2088)。

|
|
Table 4 化合物38~45的结构、物化性质和抗疟活性。[a]P. berghei和P. yoelii感染小鼠后当天皮下注射或灌胃给药, 第4天测定外周血中的原虫计数, 计算ED90。[b]ED90(青蒿琥酯)/ED90(受试化合物), 数值越高表示活性越强。[c]对照药青蒿琥酯的活性在不同的实验中有变异 |
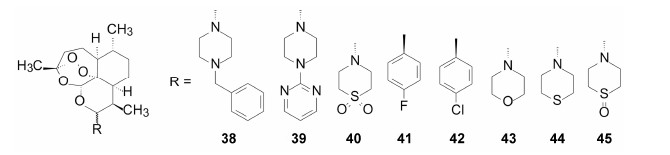
表 4数据提示,化合物38、39、41、43和44的活性很强,但体外培养显示有神经毒性,而且溶解性较差。41和42引起小鼠不协调的步态。而40有良好的物化和药理性质,进而大动物实验证实其安全和有效,遂命名为青蒿酮 (artemisone) 进入临床研究,现处于临床II期。
6 结语青蒿素的发现和相关药物的发明,开创了治疗疟疾的崭新领域。我国科学家发明的青蒿素类的单药和复方制剂,成为国际上治疗恶性疟的标准药物,已拯救了数以百万计患者的生命。青蒿素的发现者屠呦呦因此获得了2015年诺贝尔医学或生理学奖。
青蒿素类药物被世界认可和巨大成功,是当年全国“523任务”组织了近千名化学、生药学、药物化学、药理学、毒理学、药剂学、临床医学、工艺研究和工业部门等科学技术人员的劳动成果,也与负责523任务的组织和行政管理 (如张剑方、周克鼎等) 的辛勤工作分不开的。研发过程是在“文革”的特定环境、物力和技术装备极其匮乏的条件下完成更显得不易。这种特殊的组织和实施模式在当今未必有示范和再现性,而且在知识产权保护和市场销售的份额上也留下了诸多遗憾,但无论如何这个对人类有重大贡献的研发历程,应当有个客观的科学评述。限于公开的原始资料的不足,本文只是从药物化学的视角简要地叙述了青蒿素类药物的发现发明过程。所幸当年的主要贡献者分别撰写的著作,再现了当时的全息过程与场景,读者可以深入研究。重要的专著有: ① 李国桥,李英,李泽琳,曾美怡等编著. 青蒿素类抗疟药. 科学出版社,2015年,北京. ② 刘旭主编. 青蒿琥酯的研究与开发. 漓江出版社,2010年,桂林. ③ 屠呦呦编著. 青蒿和青蒿素类药物. 化学工业出版社,2006年,北京. ④ 张剑方著. 迟到的报告—五二三项目与青蒿素研发纪实. 羊城晚报出版社,2006年,广州.
具有特定结构骨架的青蒿素类药物,尚有巨大的研究空间: 其作用靶标和抗疟机制有待深入揭示; 改善药代和寻找针对耐受青蒿素原虫的新一代药物尚须继续努力。此外青蒿素的作用杂泛性也为研发 其他治疗领域的药物提供了线索 (Crespo-Ortiz MP,Wei MQ. Antitumor activity of artemisinin and its derivatives: from a well-known antimalarial agent to a potential anticancer drug. J Biomed Biotechnol,2012: Article ID 247597; Wang JX,Tang W,Zuo JP. The anti-inflammatory and imunosuppressive activity of artemisinin derivatives. Int J Pharm Res,2007,4: 336-340; 李洪军,汪伟,梁幼生. 双氢青蒿素抗寄生虫作用研究进展. 中国血吸虫病防治杂志,2011,23: 460-464)。