 2015, Vol. 50
2015, Vol. 50

2. 江苏省炎症与分子药靶重点实验室, 江苏 南通 226001;
3. 中国药科大学生命与科学技术学院, 江苏 南京 210009;
4. 南京师范大学附属中学, 江苏 南京 210009

 , WANG Xiao-li1,2, WANG Hui3
, WANG Xiao-li1,2, WANG Hui3
2. The Key Laboratory of Inflammation and Molecular Drug Target of Jiangsu Province, Nantong 226001, China;
3. College of Life Science and Technology, China Pharmaceutical University, Nanjing 210009, China;
4. The High School Affiliated to Nanjing Normal University, Nanjing 210009, China
对人类健康而言,细菌感染一直是持久而不断增长的威胁。近年来,由于人们不断地使用和滥用抗菌药物来治疗各种感染性疾病[1, 2, 3],导致许多新类型的耐药病原体出现,尤其是耐甲氧西林金葡菌和抗肺炎链球菌[4]。研究表明,MRSA等革兰阳性菌正发展为多种抗生素耐药菌: 耐青霉素类 (苯甲异噁唑青霉素和氨苄青霉素)、耐万古霉素类和耐氟喹诺酮类等药物细菌[5, 6]。因此,发展新的抗生素来对抗这些耐药菌株已经成为科学家紧迫需要解决的问题。
截短侧耳素 (1) (图 1) 是在两种天然担子菌 (Pleurotus mutilus、Pleurotus passeckerianus) 中分离出来的一种化合物,带有刚性的5-6-8三环二萜结构碳骨架的八元环[7, 8]。研究认为,它显示出对革兰阳性菌和支原体的高活性抑制作用,与它能选择性地抑制细菌蛋白质合成、与原核生物的核糖体的50s亚单位相互作用有关。但真核生物核糖体不含50s亚单位,因此对哺乳动物核糖体蛋白合成没有作用,避免了对人体细胞核糖体蛋白合成的干扰,同时因作用机制独特,与市场上现有销售药物也没有交叉耐药性[9, 10]。对截短侧耳素化合物的修饰主要集中在C-14乙醇酸链上。两个截短侧耳素衍生物泰妙菌素2和 沃尼妙林3 (图 1) 也已经成功开发为上市兽用抗感染药物[7, 9, 11]。沃尼妙林3曾被试验性地使用在罕见的疾病案例中,但因它对患者毒性太大而没有被批准上市[12]。后来又设计了人用衍生物阿扎莫林4 (图 1),成功进入了一期临床试验,因其治疗创伤性皮肤感染时只有较低的溶解度和较短的体内半衰期,最终停止了对其进一步研究[13]。第一个人用截短侧耳素衍生物是瑞他帕林 (5),它在体外显示了很强的抗菌活性且在2007年4月被FDA作为外用药正式批准 上市,主要用于脓疱病的局部治疗和创伤性皮肤二次感染[14]。近几年,其他几个新型截短侧耳素衍生物BC-3205 (6)、BC-3781 (7) 和BC-7013 (8) (图 1) 也进入了人类临床研究[7, 15, 16]。尤其是BC-3781临床治疗复杂的二期皮肤感染时,显示了良好的耐受性[17]。虽然最近人们不断设计合成了很多新型截短侧耳素衍生物用于治疗传染性病原体感染[7, 9, 10, 11, 18, 19, 20, 21],但迄今为止,人用口服的截短侧耳素衍生物药物依然尚未上市。本课题组前期研究结果表明,在C-14号侧链上含有2-氨基噻唑的截短侧耳素衍生物WL001 (9),对一些革兰阳性耐药菌的活性抑制强于阿莫西林和泰妙菌素[22, 23]。为此,决定使用WL001作为先导化合物进一步设计合成新的截短侧耳素衍生物,并评估它们的抗菌生物活性。

|
Figure 1 Pleuromutilin derivatives |
在药物化学研究中,利用含氮杂环的结构片段能很好地减少活性分子的疏水性,从而提高生物利用度,改变分子电荷分布是化合物修饰与结构改造的一种重要手段[24],所以许多具有重要生物活性的化合物和临床药物都具有含氮杂环结构片段[25]。基于这种思路,通过在截短侧耳素衍生物WL001的2-氨基噻唑基团上连接不同的2-乙酰氨取代基作为极性和水溶性片段,设计合成了系列新的化合物,以改善药物溶解度,提高分子生物利用度,改善药物代谢稳定性,并对目标化合物进行了抗耐药菌活性的体外评估。
先导化合物WL001和目标化合物15a~15j的合成如合成路线1所示,首先在无水乙醇溶液里,1,3-二氯丙酮10与硫脲11在0 ℃反应2 h,得到2-氨基噻唑-4-甲基-S-异硫脲双盐酸盐12,产率72%; 随后,化合物12的水溶液于0 ℃~室温、在25%NaOH水溶液催化下,与14-去氧-(2-碘乙酸酯)-截短侧耳素衍生物13的THF溶液发生亲核反应2 h,得到截短侧 耳素衍生物WL001 (9),产率81%; 然后,在CH2Cl2溶液里,化合物9在K2CO3存在下,于0 ℃~室温下,进一步与2-氯乙酰氯反应1.5 h,得到中间产物氯乙酸酯衍生物14,产率83%; 最后,化合物14在KI和K2CO3催化下,于四氢呋喃溶液中与含氮杂环反应,得到53%~56% 的目标产物15a~15j。目标产物均用柱色谱纯化,其结构通过IR、1H NMR和13C NMR、MS和元素分析来确定。
结果与讨论 1 化合物的合成化合物12极易溶于水,而化合物13碘化物则是脂溶性化合物,两者之间亲核取代反应在强碱NaOH水溶液催化下进行。因此在本反应未加入相转移催化剂情况下,先将化合物12的饱和水溶液和碘化物13的THF溶液混合,然后将高浓度25% NaOH水溶液缓慢滴入,这样可最大程度提高中间体WL001产率。化合物9与氯乙酰氯反应是由酰氯形成酰胺反应,根据反应机制,维持反应低温与中性偏碱的pH值是必要的。氯代化合物14的氨基化反应也是在弱酸强碱盐碳酸钾催化下进行。
目标化合物均按一般合成实验程序合成 (化学合成部分),均为白色泡沫固体,产率与ESI-MS数据及元素分析数据见表 1,NMR和红外光谱数据见表 2。
|
|
Table 1 The yields,ESI-MS data and elemental data of 15a-15j |


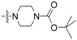
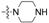

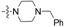


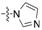

|
|
Table 2 The NMR (CDCl3/TMS) and IR spectra data of 15a-15j |

|
Scheme 1 Synthetic route of target compounds. Reaction conditions and reagents: a) Anhydrous ethanol,dry CH2Cl2,0 ℃,2 h,72%; b) 25% NaOH aqueous solution-THF,0 ℃-r.t.,2 h,81%; c) 2-Chloroacetyl chloride,K2CO3,CH2Cl2,0 ℃-r.t.,1.5 h,83%; d) Nitrogen- containingheterocycles,K2CO3,THF,40 ℃,4-8 h,53%-65% |
表 3和表 4显示了合成的化合物及对比阳性药物阿莫西林、泰妙菌素、左氧氟沙星和WL001的最小抑菌浓度。
如表 3所列数据显示,大多数合成的化合物均有很高的抑菌活性,对革兰阴性菌和革兰阳性菌的抑制能力一般近于或者高于对照阳性药物阿莫西林、泰妙菌素和左氧氟沙星。在表 4中,目标化合物对革兰阳性菌的抑菌活性显示出了更好的抗菌活性,而且对耐药细菌抑制活性优于对敏感细菌的抑制活性。
|
|
Table 3 Antibacterial activity (MIC/μg·mL-1) of pleuromutilin derivatives 15a-15j in vitro. MIC: Lowest concentration of compound that inhibits visible growth of the organism. aAll of strains were isolated from the clinical bacteria in the Nanjing Gulou hospital and reserved in Department of Microbiology,College of Life Science and Technology,China Pharmaceutical University. Clinical strains were identified by API bacteria analysis system. MSSA: Methicillin-sensitiveStaphylococcus aureus; MRSA: Methicillin-resistantStaphylococcus aureus; MSSE: Methicillin-sensitivity Staphylococcus epidermidis |
|
|
Table 4 Antibacterial activity (MIC/μg·mL-1) of pleuromutilin derivatives 15a-15j in vitro. MRSE: Methicillin-resistant Staphylococcus epidermidis; PSSP: Penicillin-susceptibleStreptococcus pneumoniae; PRSP: Penicillin resistant Streptococcus pneumonia; MC: Moraxella catarrhalis |
特别是带有哌啶或吗啉杂环片段的化合物15a和15b,以0.062 5~0.5 μg·mL-1的最低抑菌浓度,显示出了最强的抑制耐甲氧西林金葡菌 (MRSA) 和耐甲氧西林表皮葡萄球菌 (MSSA) 生物活性的能 力,几乎是先导化合物WL001的1~2倍 (0.062 5~0.5 μg·mL-1),是阿莫西林 (MIC = 0.125~8 μg·mL-1) 和泰妙菌株 (MIC = 0.062 5~2 μg·mL-1) 的8~16倍。此外,化合物15a和15b对于2种在临床上采集的 革兰阴性菌卡他拉莫菌菌株 (MC),还显示了与WL001相当的抑制活性,明显优于阿莫西林、泰妙菌素和左氧氟沙星。相反,带有杂环片段的截短侧耳素衍生物15h~15j显示了比其他化合物更低的抑菌活性。结果表明,不同的杂环化合物对2-氨基噻唑短 侧耳素衍生物的侧链修饰在影响其衍生物的抗菌活性方面发挥着较为重要的作用,并且具有哌啶或吗啉杂环的WL001确实比原型化合物有更高的抑菌活性。
通过对在噻唑侧链上带有不同2-(取代杂环化合物)乙酰胺基基团的截短侧耳素衍生物15a~15j进行构效关系分析 (SAR),结果发现,它们对革兰阳性和革兰阴性菌株的抑菌活性有很大差别。含有饱和氮杂环的化合物15a~15g比含有不饱和氮杂环的化合物15h~15j对所有受试细菌具有更显著的抑菌活性。而在饱和杂环取代的化合物中,含有哌嗪的化合物15c~15g显示中等的抑菌活性,但相对于有哌啶或吗啉的化合物15a和15b而言,它们的抗菌活性略弱。此外,带有单哌嗪环结构的N-取代哌嗪或碎片化合物15c~15g均不能有效地改善他们的抗菌活性。含有各种杂环的不同截短侧耳素衍生物对受试细菌的抗菌活性顺序如下: 哌啶 ≥ 吗啉> N-羟乙基哌嗪 > 哌嗪 ≥ N-甲基哌嗪 > N-Boc-哌嗪 >N-苄基哌嗪 > 咪唑或苯三唑。将化合物15a和15b与文献中含有不同杂环的其他截短侧耳素衍生物进行了抑菌活性比较,这些含有哌啶和吗啉环的新型氨基噻唑的系列衍生物表现出了良好的抗菌活性,其抗菌活性与带有嘌呤片段的截短侧耳素的衍生物的抗菌活性相当[20],但优于带有吡唑类片段的截短侧耳素衍生物的抗菌活性[26]。以上所述仅是初步研究结论,对于这些截短侧耳素衍生物构效关系的进一步研究还在进行中。
3 小结通过先导化合物WL001的2-氨基噻唑环与不同的2-乙酰胺杂环取代侧链连接,合成了系列新型的截短侧耳素衍生物,并初步评估他们在体外对革兰阳性和革兰阴性菌的抑制活性。结果发现大部分化合物不仅对药物敏感菌株有很高的抑制活性,同样对耐药菌株也有相同效果。特别值得注意的是,化合物15a和15b对4种MRSA菌株和MRSE菌株都表现出强大的抗菌性 (0.062 5~0.5 μg·mL-1),明显优于阿莫西林、泰妙霉素或左氧氟沙星。此外,15a和15b也能有效地抑制革兰阴性菌卡他莫拉菌。这些结果可能会提供新颖的截短侧耳素的设计思路,并为进一步研究人类临床用抗生素奠定基础。
实验部分XT-4型显微熔点测定仪 (温度未校正,北京泰克仪器有限公司); Nicolet iS10红外光谱仪 (Nicolet FTIR IS10,美国Thermo Fisher Scientific公司); Bruker Avance核磁共振仪400 MHz (瑞士Bruker BioSpin公司,TMS为内标); Shimadzu GC-MS 2010 (EI) (日本岛津公司) 和Mariner Mass Spectrum (ESI) 质谱仪 (美国 Applied Biosystems公司); FlashEA 1112元素分析仪 (美国Thermo Fisher Scientific公司)。所有实验使用溶剂均为分析纯。
抗菌实验所用菌种由南京鼓楼医院2010~2012年临床分离并提供,并由全自动细菌鉴定及分析系统VITEK确定到种,保存于中国药科大学微生物学教研室。化合物体外抗菌试验共使用10种革兰阳性耐药菌菌株和8种革兰阳性敏感菌菌株和2种革兰阴性菌菌株,包括3种甲氧西林敏感金葡菌 (Methicillin- sensitive Staphylococcus aureus,MSSA)、4种耐甲氧西林的金葡菌菌株 (Methicillin-resistant Staphylococcus aureus,MRSA)、3种表皮葡球菌敏感菌株 (Methicillin- sensitivity Staphylococcus epidermidis,MSSE)、4种耐甲氧西林表皮金葡菌 (Methicillin-resistant Staphylococcus epidermidis,MRSE)、2种青霉素敏感肺炎链 球菌 (Penicillin-susceptible Streptococcus pneumoniae,PSSP)、2种耐青霉素肺炎链球菌 (Penicillin resistant Streptococcus pneumonia,PRSP) 和2种卡他莫拉菌 (Moraxella catarrhalis,MC)。
抗菌试验所用原料截短侧耳素1购于盐城科菲特生化技术有限公司,阳性对照化合物阿莫西林购自珠海联邦制药股份有限公司,泰妙霉素购自南通市赛奥生化有限公司,左氧氟沙星购自哈药集团三精制药厂。
1 化学合成 1.1 2-氨基-噻唑基-4-甲基-S-异硫脲基双盐酸盐 (12)将硫脲11 (1.82 g,24 mmol) 完全溶解于50 mL无水乙醇溶液后,在0 ℃下缓慢加入溶于含有1,3-二氯丙酮 (1.50 g,12 mmol) 的30 mL干燥CH2Cl2溶液。在冰浴条件下,搅拌反应2 h。反应结束后将固态物滤出,并用30 mL CH2Cl2清洗,再用乙醇/乙酸乙酯混合溶液重结晶,获得2-氨基噻唑-4-甲基- S-异硫脲双盐酸盐 (2.76 g,72%),mp 89~91 ℃,与文献报道数据一致[27]。
1.2 14-O-[2-(2-氨基-噻唑基-4-甲基)硫乙酰基]木替林 (WL001,9)将2-氨基噻唑-4甲基-S-异硫脲双盐酸盐12 (2.87 g,11 mmol) 溶于5 mL水溶液中,冷至0 ℃,搅拌下将化合物12的水溶液和12 mL 25% NaOH溶液同时缓慢滴入溶于50 mL四氢呋喃的14-去氧-(2-碘乙酸酯)-木替林13 (5.00 g,10 mmol)[28]溶液中。反应在0~20 ℃条件下混合搅拌2 h。反应完毕,用旋转蒸发仪除去反应溶剂,将获得残液用40 mL乙酸乙酯溶剂萃取,并用饱和氯化钠溶液洗涤2次,用无水硫酸镁干燥乙酸乙酯溶液,过滤,浓缩,用柱色谱法提纯所得浓缩物 (PE-EtOAc,5∶1~1∶1),得到WL001 9的白色固体产物 (4.10 g,81%),mp 67~70 ℃; MS m/z 507 [M+H]+,与文献报道一致[22, 23]。
1.3 14-去氧-[2-[2-(2-氯乙酰氨基)噻唑-4-甲硫基]乙酸酯]木替林(14) 将化合物9 (5.00 g,9.9 mmol) 和K2CO3 (2.05 g,14.8 mmol) 溶于80 mL干燥的CH2Cl2,0 ℃下,将溶于15 mL干燥过的CH2Cl2中的2-氯乙 酰基氯 (1.65 g,14.8 mmol) 缓慢加入其中,保持温度,搅拌1.5 h,反应完毕,反应液用水洗3次,有机层用Na2SO4干燥,分离,浓缩,用柱色谱法分离 (PE/EtOAc = 2∶1),得到白色蜡状固体 (4.77 g,83%): MS m/z 581 [M-H]-,与文献报道一致[22, 23]。
1.4 目标化合物15a~15j的一般程序化合物 14 (1.72 mmol)、含氮杂环化合物 (8.60 mmol)、KI (0.17 mmol) 和K2CO3 (1.72 mmol) 溶于20 mL乙腈,在40 ℃下搅拌4~8 h。旋转蒸发除去反应溶剂,获得油状产物溶于乙酸乙酯,并用饱和食盐水洗涤,有机层用硫酸镁干燥,分离,浓缩并用柱色谱法分离 (EtOAc-MeOH,20∶1-8∶1) 得到产物15a~15j。
2 体外抗菌活性实验[29]采用琼脂平板稀释法,以阿莫西林、泰妙菌素和左氧氟沙星及WL001作为阳性对照药物,测定截短侧耳素的衍生物15a~15j的体外抗菌活性。
将已倍比稀释的截短侧耳素衍生物加入融化并冷却至50 ℃左右的定量MH琼脂中,制成含不同递减浓度 (0.031 25~32 μg·mL-1) 的截短侧耳素衍生物的琼脂平板,然后将在牛肉汤里过夜培养的上述受试细菌溶液,用多点接种器接种至平板,每点含菌1×105 CFU,37 ℃孵育18~20 h,测定MIC。肉眼观察细菌生长,以未见细菌生长的平板的最低药物浓度为最低抑菌浓度 (MIC)。
| [1] | Xiao YH, Giske CG, Wei ZQ, et al. Epidemiology and characteristics of antimicrobial resistance in China [J]. Drug Resist Update, 2011, 14: 236-250. |
| [2] | Livermore DM. Bacterial resistance: origins, epidemiology, and impact [J]. J Clin Infect Dis, 2003, 36: S11-23. |
| [3] | Xu ZQ, Xu ZY. Recent progress in development of antibiotics against Gram-negative bacteria [J]. Acta Pharm Sin (药学学报), 2013, 48: 993-1004 |
| [4] | Hanberger H, Walther S, Leone M, et al. Increased mortality associated with methicillin-resistant Staphylococcus aureus (MRSA) infection in the intensive care unit: results from the EPIC II study [J]. Int J Antimicrob Agents, 2011, 38: 331-355. |
| [5] | Hidron AI, Edwards JR, Patel J, et al. NHSN annual update: antimicrobial resistant pathogens associated with healthcare- associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007 [J]. Infect Control Hosp Epidemiol, 2008, 29: 996-1011. |
| [6] | Rozen DE, McGee L, Levin BR, et al. Fitness costs of fluoroquinolone resistance in Streptococcus pneumoniae [J]. Antimicrob Agents Chemother, 2007, 51: 412-416. |
| [7] | Novak R, Shlaes DM. The pleuromutilin antibiotics: a new class for human use [J]. Curr Opin Investig Drugs, 2010, 11: 182-191. |
| [8] | Fazakerley NJ, Helm MD, Procter DJ. Total synthesis of (+)- pleuromutilin [J]. Chemistry, 2013, 19: 6718-6723. |
| [9] | Mao D, Cai ZY, Zhou WC. Progress in research of mutilin antibacterials [J]. J Chin Med Ind (中国医药工业杂志), 2010, 41: 374-383. |
| [10] | Novak R. Are pleuromutilin antibiotics finally fit for human use? [J]. Ann N Y Acad Sci, 2011, 1241: 71-81. |
| [11] | Tang YZ, Liu YH, Chen JX. Pleuromutilin and its derivatives -the lead compounds for novel antibiotics [J]. Mini-Rev Med Chem, 2012, 12: 53-61. |
| [12] | Heilmann C, Jensen L, Jensen JS, et al. Treatment of resistant mycoplasma infection in immunocompromised patients with a new pleuromutilin antibiotic [J]. J Infect, 2001, 43: 234-238. |
| [13] | Phillips OA, Sharaf LH. Pleuromutilin antibacterial agents: patent review 2001-2006 [J]. Expert Opin Ther Pat, 2007, 17: 429-435. |
| [14] | Dhingra D, Parakh A, Ramachandran S. Retapamulin: a newer topical antibiotic [J]. J Postgrad Med, 2013, 59: 127-130. |
| [15] | Ross JE, Sader HS, Ivezic-Schoenfeld Z, et al. Disk diffusion and MIC quality control ranges for BC-3205 and BC-3781, two novel pleuromutilin antibiotics [J]. J Clin Microbiol, 2012, 50: 3361-3364. |
| [16] | Sader HS, Paukner S, Ivezic-Schoenfeld Z, et al. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs) [J]. J Antimicrob Chemother, 2012, 67: 1170-1175. |
| [17] | Prince WT, Ivezic-Schoenfeld Z, Lell C, et al. Phase II clinical study of BC-3781, a pleuromutilin antibiotic, in treatment of patients with acute bacterial skin and skin structure infections [J]. Antimicrob Agents Chemother, 2013, 57: 2087-2094. |
| [18] | Dreier I, Hansen LH, Nielsen P, et al. A click chemistry approach to pleuromutilin derivatives. Part 3: extended footprinting analysis and excellent MRSA inhibition for a derivative with an adenine phenyl side chain [J]. Bioorg Med Chem Lett, 2014, 24: 1043-1046. |
| [19] | Dong YJ, Meng ZH, Mi YQ, et al. Synthesis of novel pleuromutilin derivatives. Part 1: preliminary studies of antituberculosis activity [J]. Bioorg Med Chem Lett, 2015, 25: 1799-1803. |
| [20] | Ling CY, Fu LQ, Gao S, et al. Design, synthesis, and structure- activity relationship studies of novel thioether pleuromutilin derivatives as potent antibacterial agents [J]. J Med Chem, 2014, 57: 4772-4795. |
| [21] | Shang RF, Wang SY, Xu XM, et al. Chemical synthesis and biological activities of novel pleuromutilin derivatives with substituted amino moiety [J]. PLoS One, 2013, 8: e82595. |
| [22] | Wang XY, Ling Y, Wang H, et al. Novel pleuromutilin derivatives as antibacterial agents: synthesis, biological evaluation and molecular docking studies [J]. Bioorg Med Chem Lett, 2012, 22: 6166-6172. |
| [23] | Ling Y, Wang XY, Wang H, et al. Design, synthesis, and antibacterial activity of novel pleuromutilin derivatives bearing an amino thiazolyl ring [J]. Arch Pharm (Weinheim), 2012, 345: 638-646. |
| [24] | St Jean DJ Jr, Fotsch C. Mitigating heterocycle metabolism in drug discovery [J]. J Med Chem, 2012, 55: 6002-6020. |
| [25] | Stobberingh EE. In vitro effect of YTR (tazobactam) on plasmid and chromosomally mediated beta-lactamases [J]. Chemotherapy, 1990, 36: 209-214. |
| [26] | Xu P, Zhang YY, Sun YX, et al. Novel pleuromutilin derivatives with excellent antibacterial activity against Staphylococcus aureus [J]. Chem Biol Drug Des, 2009, 73: 655-660. |
| [27] | Spracue JM, Land AH, Ziegler C. Derivatives of 2-amino-4- methylthiazole [J]. J Am Chem Soc, 1946, 68: 2155-2159. |
| [28] | Riedl K. Studies on pleuromutilin and some of its derivatives [J]. J Antibiot (Tokyo), 1976, 29: 132-139. |
| [29] | Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Approved Standard M7–A6 [S]. Wayne: Clinical and Laboratory Standards Institute, 2003. |