 2015, Vol. 50
2015, Vol. 50

阿片受体是G蛋白偶联受体, 主要分为δ、μ和κ等3类亚型, 参与镇痛、抑制肠胃蠕动、呼吸抑制、心肌保护和免疫反应等生理活动。内源性配体(例如内啡肽)或药物(如吗啡和洛哌丁胺)作用于不同的受体, 调节对疼痛的感觉和胃肠道的蠕动。研究表明, δ和μ受体在结构和功能上可发生相互作用, 形成异源二聚体, 药物若同时调节这两个受体的活性可产生有益的效果, 特别是激动肠道μ受体又同时抑制肠道δ受体, 可成为治疗胃肠道功能紊乱的有价值药物。
2 苗头化合物—自内啡肽简化而来
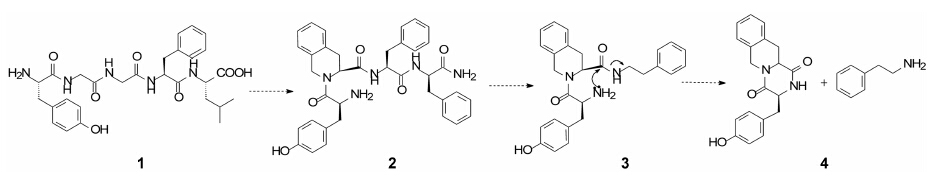
内啡肽(1, enkephalin)是阿片受体的一个重要配体, 依卢多林的研制是以这个五肽Tyr-Gly-Gly-Phe- Leu为起点的, 首先剪切掉Leu成四肽并改构氨基酸为四氢异喹啉(Tic)成化合物2(Tyr-Tic-Phe-Phe), 2仍保持1的活性。进而简化为拟二肽(3), 不仅减少了肽的性质, 还提高了对δ受体的活性, 提示四氢异喹啉的构象限制和简化成拟二肽的酰胺是个有效途径(Schiller PW, Nguyen TM, Weltrowska G, et al. Proc Natl Acad Sci USA, 1992, 89: 11871−11875)。然而3的化学稳定性低, 因为3的游离氨基对四氢异 喹啉的酰基作分子内亲核进攻, 生成取代的哌嗪二酮(4)而失去活性, 这个现象在强生公司研制拟缩胆囊素(CCK)抑制剂(也是含有游离氨基的拟二肽)时对这种环合失效进行过深入的研究(Marsden BJ, Nguyen TM, Schiller PW. Int J Pept Protein Res, 1993, 41: 313−316)。
为了避免该环合裂解反应的发生, 将四氢异喹啉的酰氨基用咪唑环替换, 以维持酰氨基的平面结构, 还保持了极性原子的分布, 这个骨架迁越是结构变换的一个重要步骤。为此合成了一系列含有咪唑基的四氢异喹啉化合物, 列于表 1中的5~10是有代表性的化合物。 3 活性评价
评价受试化合物与δ受体结合性能, 是用不同 浓度的受试物影响大鼠前脑匀浆离心制备的颗粒与放射性配体[3H]DPDPE([3H]D-Phe2, D-Phe5-内啡肽)的结合程度,测定3H的放射性活性的变化, 计算出Ki值。受试物对μ受体的Ki值用类似的方法测定, 所用放射性配体是[3H]DAMGO(D-Ala2-MePhe4-Gly-ol5-内啡肽)。
评价受试物对δ和μ受体的功能实验, 是用不同浓度的化合物分别刺激NG108-15和CHOhγ细胞膜与放射性配体[35S]GTPγS(鸟苷-5'-O-(3-[35S]硫代)三磷酸)结合的放射性活性,计算半数有效浓度EC50。
化合物在体内的活性用两种方法评价:一是用小鼠腹腔刺激实验评价镇痛作用,并以皮下注射和颅内注射两种给药途径评价化合物穿越血脑屏障的能力;另一是用小鼠玻璃球排出实验和小鼠粪便排出实验评价化合物对胃肠道功能的影响。 4 构效关系分析
分析表 1中有代表性的化合物的构效关系, 归纳如下: ① 用咪唑环替换酰胺基保持并提高了结合活性, 化合物5对δ受体的亲和力强于3, 而且也提高了选择性(比值增加), 提示酰胺向咪唑环的骨架迁越是有效果的。② 咪唑环4位被甲基或溴原子取代, 化合物6和7的活性和选择性进一步提高, 说明这个位置增加亲脂性基团有利于提高对δ受体的亲和力。③ 5位用正丙基代替苯基, 化合物8的活性显著下 降, 提示5位的苯基不宜变换。④ 母核连接咪唑的手性碳原子为S构型的活性强, 而相应的R构型的活性很弱。⑤ 将5位的苯基与咪唑并合成苯并咪唑的化合物11失去了活性。⑥ 化合物的细胞(膜)的功能性实验表明, 对δ受体的激动作用与亲和性结合作用呈平行关系。
|
|
Table 1 含咪唑基的拟二肽化合物的结构与活性 |

上述由五肽经拟二肽到咪唑基四氢异喹啉化合物的过程是苗头向先导物的过渡(hit-to-lead), 化合物5可认为是先导物, 但仍须经概念验证的体内实验。并未进行先导物优化。
用小鼠体内实验研究了化合物5的镇痛和对胃肠道蠕动的作用。实验结果表明, 外周大剂量给化合物5未显示镇痛作用, 而颅内注射有强镇痛效果, 腹腔注射显示有较强的胃肠道作用。提示5在体内可与阿片受体结合, 但难以穿越血脑屏障。
分子模拟计算表明, 化合物5的最低能量构象有两种形式: 如图 1的构象I((a))和II(b)所示, 都是由于形成了分子内氢键维持了低能构象状态。构象I的氢键给体是NH2, 氢键接受体是咪唑环的NH; 构象II的氢键给体则是咪唑环的NH, 接受体为羰基氧原子。分子力学计算表明, 构象I的稳定性强于II, 能量差值为0.5 kcal·mol−1, 量子化学计算方法也证明I为优势构象。分子模拟计算了化合物3的构象与5的构象I叠合, 表明分子的空间走向与基团的分布具有很强的适配性, 如图 1c所示(Breslin HJ, Miskowski TA, Rafferty BM, et al. J Med Chem, 2004, 47: 5000− 5020)。
 | Figure 1 化合物5的低能构象体I(a)和II(b)以及I与化合物3的叠合(c) |
化合物5下部的酪氨酸片段模拟了内啡肽的N-端基Tyr1。许多实例表明, 药物分子中含有苯酚环常因被II相代谢(如葡醛酸苷化)而呈现不利的药代性质。为此, 5的优化首先是对苯环的修饰, 合成了化合物12~29, 结构和活性列于表 2。
|
|
Table 2 化合物5和12~29的结构和活性 |
表 2的构效关系可总结如下: ① 在酪氨酸残基的苯环不同位置引入甲基, 如化合物12~14, 都不同程度地提高了对δ和μ受体的活性, 这与内源性内啡肽的N端酪氨酸被二甲基化(TMT)可提高受体结合的活性相一致(Bryant SD, Jinsmaa Y, Salvadori S, et al. Biopolymers(Pept Sci), 2003, 71: 86−102)。② 将4-羟基被其他基团取代, 无论是亲脂性或极性基团大都降低对δ和μ受体的活性。例如4位为H、F、OCH3、NH2或NHAc等化合物(17~20)的活性比相应的 羟基化合物13显著降低。③ 在咪唑环上不被甲基取代, 苯环上酚羟基用氯、氰基、甲磺酰氨基、羟甲基、乙酰基、氨磺酰基和羧基等取代的化合物, 都无活性或活性很弱。④ 酚羟基被酰胺基取代, 如化合物28仍然保持活性, 酰胺基可视作羟基的电子等排体, 在另一系列的阿片受体调节剂的研究中, 也曾有酰胺代替羟基仍保持活性的报道(Dolle RE, Machaut M, Martinez-Teipel B, et al. Bioorg Med Chem Lett, 2004, 14: 3545−3548)。甲氧基也可视作羟基的等排体(例如氢键接受体), 但化合物18无活性, 羟基既是氢键接受体也是给体, 酰胺基含有这两个因素, 反衬出活泼氢的重要性。可待因的镇痛和成瘾作用显著低于吗啡, 是由于酚羟基被甲基醚化的缘故。酰胺基取代的化合物苯环上同时被二甲基取代, 化合物29对δ和μ受体的活性明显提高, 例如对δ受体的活性提高了15倍, 对μ受体提高了近40倍, 再一次说明了含有酪氨酸片段的阿片受体调节剂二甲基化(DMT)可提高与受体的结合能力。表 2中高活性化合物进行功能性评价, 即评价化合物影响放射性配体[35S]GTPγS与细胞膜上δ和μ受体的结合能力。表 3列出了化合物的结构与数据(Breslin HJ, Cai CZ, Miskowski TA, et al.Bioorg Med Chem Lett, 2006, 16: 2505−2508)。
|
|
Table 3 有代表性的高活性化合物的功能活性 |
虽将四氢异喹啉结构改造的内容置于酪氨酸的苯环修饰之后, 但实际上与优化酚基的研究是同时或之前进行的, 原因是合成咪唑基四氢异喹啉的困难性, 为探究其他位置的构效关系, 需要付出的合成工作量太大。为简化合成, 将通式30的四氢哌啶环的两个C-C键切断, 形成通式为31的化合物。R1和R2为烷基或芳烷基, 设计R1和R2均为烷基的化合物的根据, 是四氢异喹啉用哌啶代替的一些化合物也有较强的活性。按照通式31合成的化合物及其活性列于表 4中。
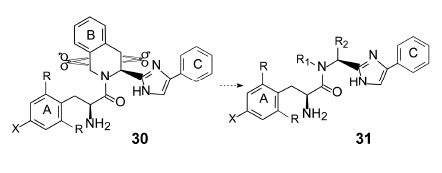
|
|
Table 4 剖列(苯并)哌啶环的化合物结构与活性 |
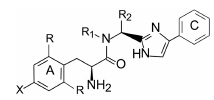


分析表 4中化合物的构效关系, 可归纳出以下的信息: ① 环A上只有羟基取代的化合物(32)活性低于有间位二甲基取代的化合物, 这与前述的四氢异喹啉系列的规律相同。新系列化合物仍以有二甲基取代为优选片段。② R1为H原子的化合物如化合物32~35与N-烷基取代的相比, 活性显著下降, 可以解释为N上氢原子可互变异构转移到酰基氧上, 形成烯醇化的羟基亚胺, 无论是反式或顺式(反式占优)都不利于活性。含有活泼氢的酰伯胺采取“假烯醇”式, 活性低于不发生互变异构的酰仲胺, 因为酰化的仲胺没有活泼氢。③ R1为烷基、R2为氢原子(该碳原子失去手性), 如化合物36和37活性显著提升, 与R1和R2都是烷基的化合物活性相近。当R2和R1分别是苄基和甲基时, 如化合物40和41, 有较强的活性。④ A环上的酚基被酰胺取代, 化合物41对δ和μ受体的活性均弱于40大约10倍。但在苯基上引入取代基, 如化合物42和43对δ和μ受体的活性都明显提高。⑤ 功能性实验意外地发现化合物42和43失去了对δ受体的激动作用, 推测是苄基苯环上连接了羧基的缘故。但43用另外的功能性实验表明对δ受体反而有拮抗作用(IC50 = 89 nmol·L−1), 而对μ受体仍保持激动作用, 尤其是化合物43引入甲氧基后, 活性比42高60倍。
化合物43对μ受体具有强激动作用(EC50 = 1 nmol·L−1), 对δ受体则为拮抗作用(IC50 = 89 nmol·L−1); 且对多种动物结肠的κ受体没有激动作用(EC50 > 1 μmol·L−1)。化合物43是拮抗δ/激动μ受体的双重调节剂, 由于胃肠道吸收很少, 因而口服给药不易进入血液循环和穿越血脑屏障, 所以降低了人体对化合物的依赖性, 这样, 43成为有潜在研发价值的候选物(Wade PR, Palmer JM, McKenney S. Br J Pharmacol, 2012, 167: 1111−1125)。 7 候选物的确定和依卢多林的上市

半体内(ex vivo)和体内(in vivo)胃肠道功能实验表明, 化合物43通过局部作用和较低的口服生物利用度作用于胃肠道上皮细胞的阿片膜受体, 因而适于作为治疗以腹泻为特征的易激性大肠炎。
43的二盐酸盐在水中的溶解度大于1 mg·mL−1, 人肝微粒体温孵的半衰期t1/2 = 150 min, 具有代谢稳定性; 对P450无抑制作用(IC50 > 20 μmol·L−1); 对hERG无抑制作用(IC50 > 10 μmol·L−1)。基于安全有效性考虑, 确定43的二盐酸盐为候选化合物, 命名为依卢多林(eluxadoline)进入开发阶段。经临床试验, 表明可治疗腹泻型肠易激综合征, 于2015年5月经FDA批准上市(Breslin HJ, Diamond CJ, Kavash RW, et al. Bioorg Med Chem Lett, 2012, 22: 4869− 4872)。