 2015, Vol. 50
2015, Vol. 50



经过近20年的发展, 高通量药物筛选 (high throughput screening, HTS) 技术已经成为新药发现的常规手段。围绕高通量筛选出现的问题也受到了广泛重视。有报告认为一个典型的高通量药物筛选模型命中率约为0.7%, 即100 000个化合物约有700个左右的活性化合物被发现[1]。但是, 这些高通量筛选模型发现的活性化合物只有少数与靶蛋白相互作用产生药效, 而多数为假阳性结果, 泛活性筛选干扰化合物 (pan-assay interference compounds, PAINS)[2]就是其中的一大类。
PAINS是一类在多种筛选模型中可以表现出阳性结果、非特异的干扰高通量筛选的化合物[3]。由于这些化合物在多种模型中都表现出活性, 很多缺乏经验的生物学家和化学家都不能识别它们。关于此类化合物药效学及作用机制的研究论文比比皆是。化学家们为了优化PAINS的活性耗费精力合成各种类似物和衍生物, 试图提高这些化合物与蛋白质之间的“亲和力”[4]。与此同时, 真正有潜力的活性化合物却被忽视。因此, 研究PAINS的产生机制以及在药物筛选中剔除PAINS的措施尤为重要。
1 PAINS的概述 1.1 PAINS概念PAINS是一类化学结构明确、在多种生物活性分析中表现活性并影响筛选结果判断又不能成药的化合物[5]。这类化合物包括多种化合物结构类型, 表现生物活性的作用机制一般与已公认的受体配体结合机制不同, 而且结合的浓度一般在微摩尔级别, 通常表现为非竞争性、无明显构效关系及较差的特异性[6]。
1.2 PAINS类型PAINS产生所谓“生物活性”主要机制有: 直接干扰筛选过程的检测信号、化合物分子形成聚合体与靶点结合[7]、产生单线态氧形成非特异性反应[8]、非特异性的蛋白残基修饰[9]及耦合金属酶发挥活性所必须的金属离子。PAINS可能包含几百种结构, 但约有数种出现频率较高的类型, 占据60% PAINS[1]。表 1 [10, 11, 12, 13, 14, 15, 16, 17]为常见PAINS的化学结构。
|
|
Table 1 |
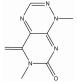
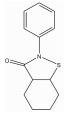
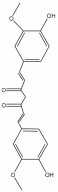
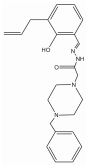
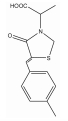
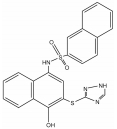
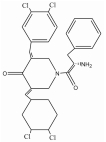
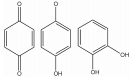
药物筛选的手段已逐渐从细胞生物学实验转变到高通量的分子生化实验。典型的生化筛选方法是测定化合物对生化反应的抑制或促进效应。这类方法的检测指标主要为光学参数, 如光的吸收值、荧光和化学发光等[18]。检测指标易于受到具有光学性质的化合物影响。
采用生物化学方法进行活性化合物筛选, 命中的化合物一般是能够与蛋白质分子相互作用, 如磷酸酶对含有磷酸基团的化合物、盐及金属等非常敏 感[18]。在检测反应荧光值的生化反应里, 荧光值很容易受荧光淬灭剂的影响[19]。亲脂性或非水溶性化合物很容易形成聚合体和沉淀干扰反应[20]。有时化合物和体系里的组分发生反应产生一些活跃的代谢产物也会影响靶点蛋白的活性。在清除反应中, 一些化合物会通过氧化DTT产生H2O2, 间接发挥了对敏感蛋白的调节作用。这些因素均可导致PAINS的产生。
2.2 化合物的化学性质活泼产生的影响一些化合物具有较活跃的化学活性, 可以与蛋白质、DNA等共价结合。这类化合物能通过不可逆反应与蛋白质结合而影响酶活性, 这类化合物被开发成为抗菌药物或抗肿瘤药物, 但这类化合物的药理活性评价主要在细胞或动物水平。一般药物与靶点的作用机制多是通过非共价相互作用调节靶蛋白功能。在分子水平, 蛋白质往往对这些含有亲电子基团的烷基、酰基化合物非常敏感, 进而引起筛选的假阳性。另外, 含有亲电子基团的假阳性化合物很容易水解或被有机溶剂分解产生活性基团, 作用于蛋白质进而引起酶活性改变。化合物成药性也取决于其活性基团的化学活性[21]。
2.3 化合物自身形成聚合体在反应体系中, 一些化合物分子间形成聚合体与靶蛋白结合。聚集体干扰蛋白活性引起假阳性结果。这类化合物主要有芳香烃类、多酚类、高亲脂类及共轭分子类化合物。
为了探究此机制, McGovern等[22]研究了14个化合物, 这些化合物在筛选中发现能抑制包含β-内酰胺酶、糜蛋白酶、二氢叶酸还原酶和半乳糖苷酶在内的多种酶活性。其抑制作用呈时间依赖性且对酶的浓度、牛血清白蛋白 (BSA) 和离子键比较敏感。在抑制过程中, 酶的变性及化合物对酶的不可逆性抑制被排除, 随着酶浓度的升高或BSA的加入, 化合物的IC50值也相应提高, 提示这些化合物通过形成聚合体非特异性的干扰酶促反应[23]。
2.4 弹头化合物弹头化合物是一类与蛋白质通过较强的非共价键 (如范德华力和金属螯合作用) 发挥相互作用的化合物。弹头化合物在体内的清除相对比较缓慢。别嘌醇是一种弹头化合物, 其发挥黄嘌呤氧化酶抑制剂活性是依赖于药物的金属螯合作用。聚离子磷酸化底物在SH2/SH3结构域与磷酰基结合位点是聚离子弹头化合物发挥作用的常见机制。总之, 弹头化合物发挥作用多依赖其化学特性而非生物学活性。
3 PAINS与高命中率化合物有些化合物在不同的筛选模型及不同的靶点激活剂/抑制剂筛选中经常被命中。其原因: ① 化合物具有多种活性, 但是对靶点的选择没有特异性, 属于多活性化合物; ② 化合物扰乱了筛选或检测方法, 形成了假阳性。PAINS通常是扰乱筛选或检测方法的一类高命中率化合物, 成药性较低, 也是影响药物研发的重要原因。
目前已经鉴定的PAINS主要有绕丹宁类、酚曼尼希碱、羟基苯腙、次烷基巴比妥酸盐和次烷基杂化化合物等。最具代表性的PAINS绕丹宁及其衍生物, 被广泛报道具有生物活性, 涉及的文献多达2 132种, 这些论文的作者来自290家研究机构[24]。绕丹宁及其衍生物在细胞及分子水平具有广泛的抗菌活性[25]。但是, 其抗菌活性的分子靶点尚未确定, 这成为进一步开发为抗菌药物的主要障碍。尽管通过靶向性设计对绕丹宁类化合物设计可以获得针对某一靶点的强有力的抑制剂, 但抗菌活性通常较弱且选择性较差[26]。通过炭疽杆菌DNA解旋酶抑制剂筛选模型获得了绕丹宁类化合物, 其IC50 0.5~4 μmol·L−1, 但其没有抗菌活性 (MIC > 100 μg·mL−1)[27]。绕丹宁类化合物也是潜在抗HIV病毒候选化合物。在化合物浓度低于微摩尔级时, 通过靶向作用于HIV病毒衣壳糖蛋白膜亚基gp41来抑制病毒对细胞的侵染。在MT2细胞水平实验中, 其能够明显抑制HIV病毒的复制, IC50为2.2 μmol·L−1 [28]。绕丹宁类化合物具有抗肿瘤活性[29], 但是也有科研工作者发现绕丹宁类化合物的细胞毒性归因于可以与细胞中多种蛋白质相互作用导致细胞死亡, 且对正常细胞也具有损伤作用。绕丹宁类化合物可以抑制TNF-α与其受体TNF receptor-1结合, 其机制是这种类型的化合物会发生光诱导反应, 从而不可逆地改变蛋白质结构。目前, 尚没有任何筛选出来的绕丹宁化合物进入了临床使用。综上, 绕丹宁相关药物的研发前景暗淡。
4 排除PAINS干扰的对策研究 4.1 生化方法PAINS在生化筛选方法中表现出令人困惑的现象, 如结构和活性的非相关性、活性呈时间相关性及陡峭的抑制曲线。为了尽可能排除因化合物分子形成聚合体而导致的酶抑制活性, 研究人员建立了一系列的标准来认定一个化合物隶属于PAINS。① 在Triton X-100存在时抑制剂活性降低[30]; ② 可以同时抑制3种以上酶的活性[31]; ③ 在抑制剂过量的前提下提高酶的浓度, 酶抑制率降低[32]; ④ 随着时间延长, 酶抑制活性提高[23]。针对化学性质活泼的化合物, 其排除可以基于化合物的化学结构, 但更为准确的筛查手段是通过它们发挥抑制作用的方式。许多所谓的抑制剂发挥酶抑制活性呈时间依赖性。酶和高浓度的抑制剂混合孵育一段时间后, 将混合液稀释至体系中抑制剂的浓度低于其IC50值, 如果抑制剂与酶的结合是不可逆的, 此时酶的活性仍被完全抑制。另外, 许多蛋白质及生物亲核物质对化学性质比较活跃的化合物很敏感, 此性质可以通过测定该类化合物对含有巯基的蛋白如谷胱甘肽的作用来测定。利用质谱分析法检测酶的分子量变化, 或利用X线衍射分析酶结构的差异[21]也可以评价化合物的化学活性, 应意识到许多化学性质活跃基团的活力又受到浓度、反应体系的pH以及靶点蛋白的特性影响[18]。
4.2 计算机预测在筛查化学性质比较活跃的化合物方面, 计算机预测是目前较为成功的技术, 其主要依据的工具为官能团过滤器[33]。Vertex 公司的REOS (rapid elimination of swill) 过滤系统能发现并清除数据库中毒性、反应性或其他方面不符合要求的化合物。过滤依据是将化合物的性质与一些不适合化合物成药性的原则相结合。REOS允许用户能定量分析每个官能团所扮演的角色, 而不是简单地将化合物移除。利用递归划分 (recursive partitioning, RP) 分析构建一个模型用以区分聚合体和非聚合体。RP可以分析一个依赖性变量 (如生物活性和分子聚合) 与不同物理化学描述符的关系[34]。
在化合物库中, 存在很多PAINS干扰药物筛选。为了排除此类化合物, Roche等[35]设计了一个虚拟筛选方法。首先, 构建了一个数据库, 由已知的至少在8个筛选模型上发挥作用的高命中率化合物、至少被6个科研项目所研究的化合物及来自于罗氏的部分化合物共同组成。通过对这些化合物结构分析并进行打分。通过该虚拟模型, 可以将90% 以上的高命中率化合物及91% 非高命中率化合物区分。该模型不是为了去除PAINS化合物, 而是使科研工作者在购买或设计化合物时尽量排除无成药可能的化合物的影响。
5 多靶点药物的研发与PAINS干扰近年来, 科学家通过对现有药物作用靶点和作用机制的研究证明, 药物发挥有效治疗疾病的药理作用通常不是单靶点的作用, 药物治疗学研究也证明, 对疾病的治疗单纯依靠单靶点的作用是不够的。因此, 多靶点药物的研究应运而生。多靶点药物是多向药理学的基础[36]。多靶点药物治疗, 简而言之, 可以同时作用于疾病网络中的多个靶点, 对各靶点的作用可以产生协同效应, 使总效应大于各单效应之和, 达到最佳的治疗效果[37]。那么, 多靶点药物应该具备什么样的条件才能成为药物? 如何区别多靶点药物与PAINS ? 根据本实验室长期进行药物筛选和活性化合物研究的结果分析, 以下方面需要重点考虑。
5.1 疾病相关靶点的选择性一种疾病涉及到的靶点是多样的但也是有限的, 而药物只有作用于这些靶点, 才可以发挥治疗相关疾病的药理作用。对于机体而言, 与疾病相关的药物靶点才是药物治疗的靶点, 药物只有与这些靶点相互作用, 才能发挥治疗作用。对于这些靶点, 表现为多靶点的特点, 作用于这些靶点的药物可以称为多靶点药物。而对于整体而言, 虽然作用是多靶点的, 但也具有明显的选择性, 针对疾病相关靶点的选择性是多靶点药物的重要成药条件之一。
本实验室在采用高通量筛选技术进行中药复方研究过程中, 根据药物作用的多靶点特点和要求, 提出了有效成分组的研究思路, 其重点内容之一就是疾病相关的多靶点组合[38]。对于单一化合物或组合化合物的多靶点作用, 这种多靶点的选择性同样是重要的。当然, 这种选择性并非要求作用于所有疾病相关靶点或无关靶点一概无作用, 选择性也是相对的。
5.2 多靶点的相关性对于多靶点的药物[39], 首先要区分多靶点的选择性, 而在具备疾病相关靶点选择性的基础上, 需要分析这些具有活性的多靶点之间的相互关系。对机体和疾病过程而言, 靶点之间并不是孤立的, 是相互作用和相互影响的。对于药物的作用, 其能够作用的靶点相互之间的影响应该是与疾病具有相关性, 或相互协调、相互促进, 共同发挥防治疾病的作用。这种相互作用可以用网络药理学的方式来解释, 最终的目的是药物作用的多靶点, 能发挥相互协调的防治疾病的作用[40]。
本实验室提出基于网点式高通量药物筛选模式, 应用多种新技术和新方法建立了功能性多靶点筛选模型, 发现作用于功能性整合药物靶点的具有自主知识产权的Ⅰ类抗脑缺血新药——匹诺塞林, 证明匹诺塞林能扩张缺血区微血管、抑制炎症反应、保护线粒体功能和抑制细胞凋亡等多方面的作用, 通过调控神经血管单元信号转导网络保护神经血管单元的形态和功能而发挥防治作用[41]。
6 总结PAINS概念的提出是药物化学和药物发现历史的重要事件。目前, 一个典型的药物筛选数据库中往往有5%~12% 化合物属于PAINS[24]。在药物筛选时应仔细研究筛选的方法, 减少PAINS的干扰。在药物研发的早期阶段, 应该利用包含计算机在内的多种方式排除PAINS[42]。对于一些明显异常的数据, 须进行不同原理的实验加以验证。通过提高技术水平, 应用合理的技术方法, 在进行HTS药物筛选时综合考虑药物成药性准则, 提高新药筛选的效率和成功率, 推动新药研发。值得注意的是, 鉴于生物系统的复杂性和药物与机体相互作用的特点, 在预测化合物成药性的过程中, 准确排除PAINS仍需要加强研究。
| [1] | Baell JB. Observations on screening-based research and some concerning trends in the literature [J]. Fut Med Chem, 2010, 2: 1529-1546. |
| [2] | Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays [J]. J Med Chem, 2010, 53: 2719-2740. |
| [3] | Canny SA, Cruz Y, Southern MR, et al. PubChem promiscuity: a web resource for gathering compound promiscuity data from PubChem [J]. Bioinformatics, 2012, 28: 140-141. |
| [4] | Camp D, Davis RA, Evans-Illidge EA, et al. Guiding principles for natural product drug discovery [J]. Fut Med Chem, 2012, 4: 1067-1084. |
| [5] | Baell JB, Ferrins L, Falk H, et al. PAINS: relevance to tool compound discovery and fragment-based screening [J]. Aust J Chem, 2014, 66: 1483-1494. |
| [6] | Dahlin J, Nissink W, Strasser JM, et al. PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS [J]. J Med Chem, 2015, 58: 2091-2113. |
| [7] | Feng BY, Shoichet BK. Synergy and antagonism of promiscuous inhibition in multiple-compound mixtures [J]. J Med Chem, 2006, 49: 2151-2154. |
| [8] | Schorpp K, Rothenaigner I, Salmina E, et al. Identification of small-molecule frequent hitters from AlphaScreen high-throughput screens [J]. J Biomol Screen, 2014, 19: 715-726. |
| [9] | Metz JT, Huth JR, Hajduk PJ. Enhancement of chemical rules for predicting compound reactivity towards protein thiol groups [J]. J Comput Aid Mol Des, 2007, 21: 139-144. |
| [10] | Latuasan H, Berends W. On the origin of the toxicity of toxoflavin [J]. Biochim Biophys Acta, 1961, 52: 502-508. |
| [11] | Devos R, Guisez Y, Plaetinck G, et al. Covalent modification of the interleukin-5 receptor by isothiazolones leads to inhibition of the binding of interleukin-5 [J]. Eur J Biochem, 1994, 225: 635-640. |
| [12] | Troselj KG, Kujundzic RN. Curcumin in combined cancer therapy [J]. Curr Pharm Des, 2014, 20: 6682-6696. |
| [13] | Uppal G, Bala S, Kamboj S, et al. Therapeutic review exploring antimicrobial potential of hydrazones as promising lead [J]. Der Pharm Chem, 2011, 3: 250-268. |
| [14] | Tomašić T, Peterlin Mašič L. Rhodanine as a scaffold in drug discovery: a critical review of its biological activities and mechanisms of target modulation [J]. Expert Opin Drug Dis, 2012, 7: 549-560. |
| [15] | Soares KM, Blackmon N, Shun TY, et al. Profiling the NIH small molecule repository for compounds that generate H2O2 by redox cycling in reducing environments [J]. Assay Drug Dev Technol, 2010, 8: 152-174. |
| [16] | Willson M, Lauth N, Perie J, et al. Inhibition of glyceraldehyde-3-phosphate dehydrogenase by phosphorylated epoxides and alpha-Enones [J]. Biochemistry, 1994, 33: 214-220. |
| [17] | Seung SA, Lee JY, Lee MY, et al. The relative importance of oxidative stress versus arylation in the mechanism of quinone-induced cytotoxicity to platelets [J]. Chem Biol Interact, 1998, 113: 133-144. |
| [18] | Rishton GM. Molecular diversity in the context of leadlikeness: compound properties that enable effective biochemical screening [J]. Curr Opin Chem Biol, 2008, 12: 340-351. |
| [19] | Simeonov A, Jadhav A, Thomas CJ, et al. Fluorescence spectroscopic profiling of compound libraries [J]. J Med Chem, 2008, 51: 2363-2371. |
| [20] | Di L, Kerns EH. Biological assay challenges from compound solubility: strategies for bioassay optimization [J]. Drug Discov Today, 2006, 11: 446-451. |
| [21] | Babaoglu K, Simeonov A, Irwin JJ, et al. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against β-lactamase [J]. J Med Chem, 2008, 51: 2502- 2511. |
| [22] | McGovern SL, Caselli E, Grigorieff N, et al. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening [J]. J Med Chem, 2002, 45: 1712- 1722. |
| [23] | McGovern SL, Shoichet BK. Kinase inhibitors: not just for kinases anymore [J]. J Med Chem, 2003, 46: 1478-1483. |
| [24] | Baell J, Walters MA. Chemistry: chemical con artists foil drug discovery [J]. Nature, 2014, 513: 481. |
| [25] | Chen ZH, Zheng CJ, Sun LP, et al. Synthesis of new chalcone derivatives containing a rhodanine-3-acetic acid moiety with potential anti-bacterial activity [J]. Eur J Med Chem, 2010, 45: 5739-5743. |
| [26] | Silver LL. Challenges of antibacterial discovery [J]. Clin Microbiol Rev, 2011, 24: 71-109. |
| [27] | Aiello D, Barnes MH, Biswas EE, et al. Discovery, characterization and comparison of inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicases [J]. Bioorg Med Chem, 2009, 17: 4466-4476. |
| [28] | He XY, Zou P, Qiu J, et al. Design, synthesis and biological evaluation of 3-substituted 2, 5-dimethyl-N-(3-(1H-tetrazol-5-yl) phenyl) pyrroles as novel potential HIV-1 gp41 inhibitors [J]. Bioorg Med Chem, 2011, 19: 6726-6734. |
| [29] | Li W, Zhai X, Zhong Z, et al. Design, synthesis and evaluation of novel rhodanine-containing sorafenib analogs as potential antitumor agents [J]. Arch Pharm, 2011, 344: 349-357. |
| [30] | Ryan AJ, Gray NM, Lowe PN, et al. Effect of detergent on “promiscuous” inhibitors [J]. J Med Chem, 2003, 46: 3448- 3451. |
| [31] | Mcgovern SL, Helfand BT, Feng B, et al. A specific mechanism of nonspecific inhibition [J]. J Med Chem, 2003, 46: 4265-4272. |
| [32] | Feng BY, Shelat A, Doman TN, et al. High-throughput assays for promiscuous inhibitors [J]. Nat Chem Biol, 2005, 1: 146- 148. |
| [33] | Nadin A, Hattotuwagama C, Churcher I. Lead-oriented synthesis: a new opportunity for synthetic chemistry [J]. Angew Chem Int Ed, 2012, 51: 1114-1122. |
| [34] | Wang L, Chen L, Liu Z, et al. Predicting mTOR inhibitors with a classifier using recursive partitioning and naïve bayesian approaches [J]. PLoS One, 2014, 9: e95221. |
| [35] | Roche O, Schneider P, Zuegge J, et al. Development of a virtual screening method for identification of “frequent hitters” in compound libraries [J]. J Med Chem, 2002, 45: 137-142. |
| [36] | Hu Y, Bajorath J. What is the likelihood of an active compound to be promiscuous? Systematic assessment of compound promiscuity on the basis of PubChem confirmatory bioassay data [J]. AAPS J, 2013, 15: 808-815. |
| [37] | Blumenfeld A, Cady R. Pharmacological synergy, multi-target therapeutics——the past and the future in headache therapy? [J]. Headache: J Head Face Pain, 2012, 52: DOI: 10.1111/j.1526-4610.2012.02283.X. |
| [38] | Du GH. Overview of effective compounds group formulation in traditional Chinese medical prescription [J]. Chin Tradit Pat Med (中成药), 2002, 24: 878-880. |
| [39] | Đilović I, Rubčić M, Vrdoljak V, et al. Novel thiosemicarbazone derivatives as potential antitumor agents: synthesis, physicochemical and structural properties, DNA interactions and antiproliferative activity [J]. Bioorg Med Chem, 2008, 16: 5189-5198. |
| [40] | Du GH, Wang YH, Zhang R, et al. Multiple ingredients target is the shallow review of the mechanism of traditional Chinese medicine [J]. Mod Tradit Chin Med Mater Med——World Sci Technol (世界科学技术: 中医药现代化), 2009: 480-484. |
| [41] | Du GH, Gao M, Guang HM, et al. Exploration for development of anti-ischemic stroke drugs: the discovery of pinocembrin——a novel agent targeting the neurovascular unit based on multi-targets and network regulation [J]. Chin J Pharmacol Toxicol (中国药理学与毒理学杂志), 2012, 26: 720. |
| [42] | Hu Y, Bajorath JR. Influence of search parameters and criteria on compound selection, promiscuity, and pan assay interference characteristics [J]. J Chem Inf Model, 2014, 54: 3056-3066. |