 2015, Vol. 50
2015, Vol. 50

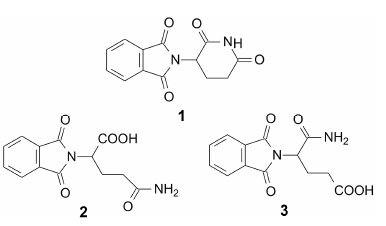
沙利度胺 (1, thalidomide) 是1957年德国Grunenthal研制的催眠、镇静和止吐的药物, 用于孕妇的妊娠反应, 恰恰由于其严重的致畸作用, 加之有外周神经炎和便秘等不良反应, 于1961年撤市。但在作为镇静药使用时, 发现沙利度胺对麻风性结节性红斑病可减轻症状, 后来还发现沙利度胺对许多疾病具有免疫调节和抗炎作用, 如风湿性关节炎、炎性大肠炎和艾滋病恶病质等。1991年证实这些抗炎作用的表现是由于抑制肿瘤坏死因子α (TNF-α) 所致。TNF-α是由免疫系统的巨噬细胞产生的细胞因子, 是炎症反应的介质。1994年又发现沙利度胺具有抗血管生成作用和抗肿瘤活性。这样多的药理活性和明确的作用靶标, 引起美国Celgene公司的兴趣, 遂以沙利度胺为先导物, 以TNF-α为靶标的抗炎活性为指标, 进行了系统深入的研究。设定的目标是通过改造沙利度胺的结构, 提高抑制TNF-α的活性, 消除致畸和其他不良反应, 提高溶解性和口服生物利用度。该公司研制的波马度胺于2013年在美国上市, 阿普度胺是沙利度胺的另一个类似物。
2 开环化合物作为结构改造的切入点
初步研究表明, 邻苯二甲酰亚胺环是重要的药效团, 开环则降低活性, 因此要保持这个环状片段不动。沙利度胺在生理条件下, 谷氨酰胺环可发生水解开环, 可能以2或3的结构与靶标结合。因而用简单的氨基酸或含取代基的氨基酸与邻苯二甲酸形成亚胺, 作为结构改造的切入点。
将甘氨酸、β-氨基丙酸或γ-氨基丁酸 (调节不同的链长) 与邻苯二甲酸酐反应生成的化合物4、5和6抑制TNF-α的活性很弱, 在数百微摩尔浓度下, 抑制率达不到50%, 提示简单的羧酸链取代没有活性。

化合物5的侧链上引入苯环, 得到的7可明显提高抑制TNF-α的活性, 与沙利度胺的活性相近 (IC50 = 260 μmol·L−1)。继之在这个苯环上进行各种取代, 活性进一步提高, 化合物8抑制TNF-α的IC50 = 5.6 μmol·L−1, 显著高于卤素、氰基或大的烷氧基取代的化合物。
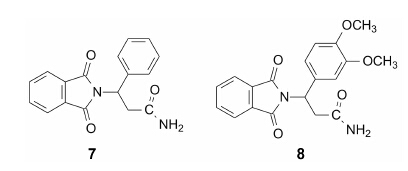
固定3, 4-二甲氧基苯基不变, 将侧链末端的CONH2用烷基酰胺CONHR、羧酸COOH、醇基CH2OH等置换, 抑制活性均有不同程度降低。然而甲酯基COOCH3的置换 (化合物9), 提高了活性, IC50 = 2.9 μmol·L−1。
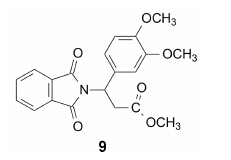
固定3, 4-二甲氧基苯基和侧链的甲酯基不变, 在邻苯二甲酰亚胺的苯环4个位置用不同的功能基取代, 例如羟基、硝基、氨基、叔丁基和氯原子取代, 活性评价结果表明, 其中的3-氨基 (10) 和4-氨基化合物 (11) 活性高于化合物9, IC50分别为0.45和0.38μmol·L−1, 这是两个里 程碑式的化合物 (Muller WG, Corral LG, Shire MG, et al. J Med Chem, 1996, 39: 3238−3240)。

3 双靶标作用: 具有抑制PDE4的沙利度胺类似物
在上述以沙利度胺为先导物的结构变换中, 虽然都程度不同地抑制TNF-α, 但进一步研究发现, 含有二烷氧苯基片段的化合物还具有抑制磷酸二酯酶4 (PDE4) 的活性, 而沙利度胺和没有该芳环的类似物 (例如来那度胺和波马度胺) 没有抑制PDE4的作用。
PDE4是环磷酸腺苷 (cAMP) 的特异性水解酶, 催化水解cAMP成AMP, 抑制PDE4的活性可提高cAMP水平, 从而影响许多细胞内的信号通路, 对炎症、呼吸道功能和中枢神经系统有重要影响。抑制PDE4是治疗炎症疾病的另一途径。实验表明, 沙利度胺类似物同时抑制PDE4的活性和TNF-α的产生, 构成抑制炎症的级联反应, 更有利于治疗与炎症相关的疾病。实验还发现这些化合物对PDE4和TNF-α的抑制活性强度并不是平行的 (Muller GW, Shire MG, Wong LM, et al. Bioorg Med Chem Lett, 1998, 8: 2669−2674; Corral LG, Kaplan G. Ann Rheum Dis, 1999,58: (Suppl I) I107−I113)。
构效关系研究表明, PDE4抑制剂的重要药效团特征之一是含有3, 4-二烷氧苯基片段, 例如PDE4 抑制剂罗力普兰 (12, rolipram) 和罗氟司特 (13, roflumilast), 而且3位的烷氧基的体积大于4位的甲氧基为宜 (Kodimuthali A, Jabaris SSL, Pal M. J MedChem, 2008, 51: 5471−5489)。因而, 下一步的结构优化的目标是同时提高对PDE4和TNF-α抑制活性。

对PDE4和TNF-α抑制活性的不平行性, 说明二者不构成因果关系, 预示进一步对侧链的优化是可能的。通式14是固定4位为甲氧基, 变换3位的烷基R为乙基或环戊基 (模仿罗力普兰), 变换侧链的末端基团Y为羧基COOH、甲酯基COOCH3、酰胺基CONH2、氰基CN和甲磺酰基SO2CH3, 结果表明, 3位的环戊基并不优于乙基, 因而确定为3-乙氧基-4-甲氧基的取代; Y的取代基中甲磺酰基对PDE4和TNF-α的抑制活性都很高, 化合物15对PDE4抑制活性IC50 = 0.22 μmol·L−1, 对TNF-α的IC50 = 1.3 μmol·L−1。
3.2 邻苯二甲酰亚胺环上取代基的优化以化合物15为新的优化起点, 考察邻苯二甲酰亚胺环上取代基对抑制PDE4和TNF-α活性的影响, 主要集中在 4位, 这或许是由于来那度胺的氨基取代基位置在4位。当4位引入羟基或甲氧基, 抑制PDE4的活性略
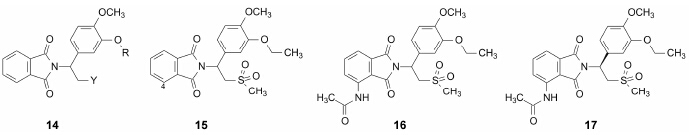
低于化合物15; 而4-CH3、4-NH2或4-N(CH3)2等化合物抑制PDE4和TNF-α的活性略强于15, 而4位为乙酰胺基的化合物16 (消旋体) 对PDE4和TNF-α抑制活性强度显著高于化合物15, IC50分别为0.082和0.19 μmol·L−1, 而且只有侧链为甲磺酰基时才有这样高的活性。
3.3 S-异构体活性强于R-异构体化合物16是消旋化合物, 是优化结构中活性最强的化合物, 分子中 含有1个手性中心, 而且该手性碳相连的氢原子不是酸性氢, 不会经互变异构而转变构型,因此可将对映异构体拆分为稳定的光活体。实验表明, S-构型(17) 对PDE4和TNF-α的抑制活性强于R-异构体5倍以上, IC50分别为0.074和0.077 μmol·L−1 (Man HW, SchaferP, Wong LM, et al. J Med Chem, 2009, 52: 1522−1524)。
4阿普斯特的批准上市
动物实验表明, S-异构体17具有良好的药代动 力学性质, 对雌性大鼠的口服生物利用度F = 64%, 半衰期t1/2= 5 h, 且具有中等程度的分布容积和低清除率, 对多种CYP氧化酶无抑制作用, 血浆蛋白结合率90%。大鼠体内药效学实验表明具有较高的抗炎活性: 用脂多糖诱导TNF-α生成的抑制作用的ED50 = 0.03mg·kg−1; 脂多糖诱导嗜中性细胞的抑制作用ED50 = 0.3 mg·kg−1。遂即将化合物17确定为候选化合物, 命名为阿普斯特(apremilast) 进入开发研究, 经三期临床研究证明, 阿普斯特是口服治疗银屑病型关节炎的有效药物 (Schafer P. Biochem Pharmacol, 2012, 83: 1583−1590), 于2014年经美国FDA批准上市。