 2015, Vol. 50
2015, Vol. 50



青蒿素是我国科学工作者1971年首次从中药黄花蒿中提取分离得到的一类具有过氧桥结构的倍半萜内酯类化合物, 临床用于恶性疟疾的治疗, 由于其药代动力学方面存在缺陷, 对其进行结构修饰得到了二氢青蒿素 (dihydroartemisinin, DHA)、蒿甲醚 (artemether)、蒿乙醚 (arteether)、青蒿琥酯 (artesunate) 等, 这些化合物都具有较好的抗疟活性。近年来, 青蒿素类化合物抗肿瘤活性的研究引起了科研人员的广泛关注。
为了提高青蒿素类化合物的抗肿瘤活性、改善其药代动力学性质、降低其毒副作用, 近年来关于其结构修饰的研究有了较大进展。青蒿素类化合物C-10位化学可改造性强, 通常是在C-10位引入不同的取代片段以研究其对抗肿瘤活性的影响, 其中包括氧、氮、硫和碳等不同原子取代的C-10位化合物。
Yang等[1]分别合成了一系列10-O-查尔酮取代 的青蒿素类化合物1 (图 1) 并测定了抗肿瘤活性, 结果表明其对人白血病HL-60细胞、小鼠白血病P388细胞及耐多柔比星的P388/Adr细胞均具有较好的 生长抑制活性, 且发现青蒿醚类化合物的活性好于青蒿酯类化合物。此后, Xie等[2]合成了10-N-取代的青蒿素−查尔酮拼合物, 并测定了这类化合物对HT-29、A549、MDA-MB-231、HeLa及H460等细胞株的抑制活性, 其中代表化合物2 (图 1) 对上述所有细胞株的IC50值均小于1 μmol·L−1。

|
Figure 1 Artemisinin derivatives |
Oh等[3]以二氢青蒿素为先导化合物, 合成了一系列10-S-取代的化合物, 并测定了其对人脐静脉内皮细胞 (HUVEC) 的生长抑制活性, 结果表明, 化合物3 (IC50 = 0.9 mmol·L−1)、4 (IC50 = 1.7 μmol·L−1) 和5 (IC50 = 1.29 μmol·L−1) (图 1) 活性较好, 抑制活性优于母体化合物青蒿素 (IC50 > 50 mmol·L−1) 和DHA (IC50 = 8.9 mmol·L−1)。
青蒿素类化合物中缩醛结构的存在, 使得该类化合物在体内代谢迅速, 生物利用度较低, 设计用碳原子代替10位的氧原子的10-碳脱氧类似物, 期望这类化合物可以具有更高的水解稳定性, 更长的半衰期和更低的毒副作用。Jung等[4]报道了一些10-碳类似物6 (图 1), 药理结果显示, 大部分化合物可以有效抑制肿瘤细胞血管生成, 其效果远远好于烟曲霉素和擦里多米德。青蒿素二聚体或三聚体的报道也越来越多, Wang等[5]利用Ugi反应合成了一系列10-C-青蒿素二聚体7 (图 1), 在BT474细胞株中表现出远远强于青蒿琥酯的细胞毒作用, 其中活性最好的化合物的IC50值仅为青蒿琥酯的1/600。
青蒿素类化合物抗肿瘤活性强、毒性低, 可以通过抑制肿瘤细胞增殖[6, 7]、诱导肿瘤细胞凋亡[8, 9]、抑制肿瘤血管生成[10, 11]、逆转肿瘤细胞多药耐药[12]等多种方式抑制肿瘤的发生发展, 最近其在抗肿瘤领域的研究十分活跃, 是一类具有深入研究价值的抗肿瘤化合物。
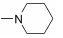
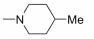
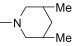
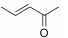
本实验室前期研究中, 以二氢青蒿素 (DHA) 为先导化合物, 对其C-10位进行结构修饰, 设计并合成了一系列青蒿素−哌嗪衍生物 (8, 9) (图 2), 并测定了这些化合物对人急性早幼粒白血病细胞HL-60、小鼠白血病P388细胞及对多柔比星耐药的P388/Adr细胞的生长抑制活性[13]。药理实验结果表明大部分哌嗪取代的青蒿素类化合物对肿瘤细胞具有较强的生长抑制活性, 并有以下结论: ① 引入取代哌嗪后化合物对HL-60、P388和P388/Adr细胞的生长抑制活性比DHA显著提高; ② 青蒿醚类化合物的抗增殖活性明显好于青蒿酯类化合物; ③ 大部分目标化合物在耐药细胞中表现出高于敏感细胞的生长抑制活性。本文在前期研究的基础上, 选择苄基、苯乙酰基和肉桂酰基为连接部分, 考察哌嗪之外的几个杂环对活性的影响, 设计如下化合物: 五元含氮杂环四氢吡咯取代的化合物GF01、ZH01、GH01; 吗啉取代物ZH02、GH02; 哌啶取代物GF05、ZH03、GH03; 唑类取代物GF02、GF03、GF04; 取代哌啶类化合物ZH04、ZH05、ZH06、ZH07和GH04。上述所设计的二氢青蒿素衍生物结构及名称见表 1。以二氢青蒿素为原料, 经三氟乙酸酐反应成为活性酯, 再与相应侧链片段反应而制得目标化合物 (合成路线1)。

|
Figure 2 Artemisinin derivatives |
|
|
Table 1 Structures and nomenclatures of dihydroartemisinin derivatives containing nitrogen heterocycles |
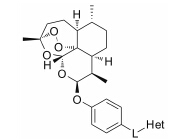

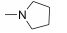



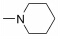


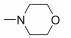


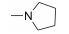

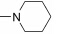

 | Scheme 1 Synthetic route of target compounds |
青蒿素类目标化合物的1H NMR、LC-MS谱图数据和熔点数据如表 2所示。根据1H NMR谱图数据, 青蒿素母核10位碳原子直接与两个氧原子相连, 10位氢原子与9位氢原子发生偶合, 其偶合常数是判断10位空间构型的重要依据: J9,10 = 3~4 Hz为β构型, J9,10 = 9~10 Hz为α构型[14], 所有化合物的C-10上氢的双重峰均出现在δ = 5.50左右, 偶合常数均为3.0 Hz, 可判断所有化合物均为β构型。GH类化合物烯烃的特征峰出现在δ = 6.85和δ = 7.43左右, 且均为双重峰, 由偶合常数15.3 Hz可判断双键为反式构型。
|
|
Table 2 The spectra data of target compounds |
采用细胞计数法测定了所有目标化合物对人急性早幼粒白血病细胞HL-60的生长抑制活性, 采用MTT法测定了所有目标化合物对人乳腺癌细胞MCF-7及耐多柔比星细胞MCF-7/Adr的生长抑制活性, 以DHA和多柔比星 (Adr) 为阳性对照, 结果见表 3。
|
|
Table 3 The antiproliferative effects of artemisinin derivatives in MCF-7 cells, MCF-7/Adr cells and HL-60 cells. DHA: Dihydroartemisinin; Adr: Doxorubicin |
采用细胞计数法和MTT法测试所合成化合物 对人急性早幼粒白血病细胞HL-60、人乳腺癌细胞MCF-7及耐多柔比星细胞MCF-7/Adr的生长抑制活性, 药理实验结果表明: ① 在敏感细胞HL-60和MCF-7中, 所有目标化合物的生长抑制活性比先导化合物二氢青蒿素都有所提升, 但弱于阳性对照药多柔比星; ② 在耐药细胞MCF-7/Adr中, 所有目标化合物表现出强于敏感细胞MCF-7的生长抑制作用, 其GI50值显著低于二氢青蒿素和多柔比星, 说明该类化合物对多药耐药肿瘤的治疗可能具有潜力; ③ 杂环取代对化合物活性的影响不显著, 也不存在明显规律性; ④ GH类化合物在敏感细胞HL-60、MCF-7和耐药细胞MCF-7/Adr中的GI50值差别不大, 较GF和ZH类化合物在敏感细胞HL-60和MCF-7的GI50值波动较小, 有可能与肉桂酰基中α, β-不饱和体系的存在有关。化合物GF02、GH04、ZH04在3个细胞株中均表现出强烈的生长抑制活性, 值得深入研究。
熔点用X-4数字显微熔点测定仪 (上海精密科学仪器有限公司) 测定, 温度未经校正。1H NMR采用Bruker ARX-400型核磁共振波谱仪测定, TMS为内标。MS美国Waters公司Waters Quattro micro API三重四极杆串联质谱仪测定。柱色谱用200~300目硅胶、薄层色谱用硅胶GF254均由青岛海洋化工厂生产。含有二氢青蒿素母核的化合物反应及后处理用水为纯净水或重蒸水, 其他试剂均为分析纯或化学纯的市售商品, 使用时根据实验需要进一步纯化。 实验部分 1 化合物的合成 1.1 中间体的合成 1.1.1 4-(1-四氢吡咯甲基) 苯酚 (FA01) 的合成
将1.22 g (0.01 mol) 对羟基苯甲醛溶于30 mL无水甲醇中, 搅拌下滴加1.25 mL (0.015 mol) 四氢吡咯, 室温搅拌30 min, 分批加入硼氢化钠0.38 g (0.01 mol), 加毕, 使用TLC (二氯甲烷−甲醇, 12∶1) 监测直至原料点消失。减压浓缩溶液, 乙醇−水重结晶得到砖红色粉末状固体, 收率78.2%。
1.1.2 4-(1-咪唑基甲基) 苯酚 (FA02) 的合成将1.24 g (0.01 mol) 对羟基苄醇和1.02 g (0.015 mol) 咪唑加入到圆底烧瓶中, 90 ℃下共融反应。自然冷却至室温, 加入30 mL乙酸乙酯, 有大量固体析出, 使用TLC (二氯甲烷−甲醇, 12∶1) 监测反应, 抽滤, 以乙酸乙酯洗涤滤饼, 红外灯下干燥, 得到白色粉末状固体, 收率82.8%。
以1, 2, 3-三氮唑和1, 2, 4-三氮唑为原料, 按照FA02合成方法, 得到FA03和FA04。
1.1.3 4-[1-(4-(2-甲氧基苯基) 哌嗪) 甲基] 苯酚 (FA05) 的合成将1.22 g (0.01 mol) 对羟基苯甲醛和1.92 g (0.01 mol) 1-(2-甲氧基苯基) 哌嗪加入到圆底烧瓶中, 60 ℃下共融。缓慢滴加0.37 mL (0.01 mol) 的甲酸, 滴加完毕后升温至90 ℃回流反应2 h, 使用TLC (二氯甲烷−甲醇, 12∶1) 监测反应, 自然冷却至室温, 加入30 mL乙酸乙酯, 有大量固体析出, 抽滤, 以乙酸乙酯洗涤滤饼, 红外灯下干燥, 得到白色粉末状固体, 收率68.3%。
1.1.4 1-(1-四氢吡咯基)-2-(4-羟基苯基)-乙酮 (ZHA01) 的合成将1.52 g (10 mol) 对羟基苯乙酸和1.012 g (10 mol) 三乙胺溶解于30 mL精制CH2Cl2中, 搅拌回流30 min。缓慢滴加0.87 mL (12 mmol) 二氯亚砜, 继续回流搅拌2 h, 自然冷却至室温。加入0.92 mL (11 mmol) 四氢吡咯, 回流状态下搅拌反应2 h, 使用TLC (二氯甲烷−丙酮, 1∶1) 监测反应。反应结束后直接用硅胶柱色谱分离 (二氯甲烷−丙酮, 5∶1~1∶1), 得浅黄色油状物, 收率46.7%。
按照ZHA01的合成方法, 以对羟基苯乙酸和相应杂环为原料, 得到中间体ZA02~ZA07, 采用相同的方法, 以对羟基肉桂酸为原料, 得到HA01~HA04。
1.2 10-O-[4-(1-四氢吡咯基甲基) 苯基]-(10S)-二氢青蒿素 (GF01) 的合成将0.568 g (2 mmol) 二氢青蒿素、0.56 g (4 mmol) 三乙胺、30 mL精制二氯甲烷于冰盐浴下搅拌30 min, 滴加0.566 g (4 mmol) 三氟乙酸酐 (TFAA), 搅拌 反应至TLC显示无DHA存在, 加入3 mmol中间体FA01, 室温下搅拌24 h。停止反应, 依次用5% NaOH溶液 (30 mL/次×3次)、30 mL H2O、30 mL饱和NaCl水溶液洗, CH2Cl2层用无水Na2SO4干燥过夜, 使用TLC (氯甲烷−丙酮, 3∶1) 监测反应。滤去干燥剂, 减压浓缩滤液, 得到黄色油状物。用硅胶柱色谱分离 (二氯甲烷−丙酮, 10∶1~5∶1), 得白色固体, 收率22.3%。
分别以FA02~FA05、ZA01~ZA07和HA01~FA05为原料, 根据GF01的合成方法, 得到GF02~GF05、ZH01~ZH07和GH01~GH04, 产率10%~20%。
2 肿瘤细胞生长抑制活性测定 2.1 细胞培养HL-60细胞培养于含有10% (v/v) 胎牛血清、100 u·mL−1青霉素、100 μg·mL−1链霉素的RPMI 1640 培养液中; 人乳腺癌细胞MCF-7及耐多柔比星细胞MCF-7/Adr培养于含有10% 胎牛血清、100 u·mL−1青霉素、100 μg·mL−1链霉素、0.37% NaHCO3的DMEM培养液中。上述细胞均置于5% CO2、饱和湿度、37 ℃培养箱内培养。
2.2 抗肿瘤活性测定将药物溶于二甲基亚砜配成8 mmol·L−1储存溶液, 用无水乙醇和培养液稀释至适当浓度与HL-60细胞共同孵育72 h。经药物处理的细胞悬液, 用血球计数板计总细胞数。按下式计算细胞生长抑制率:
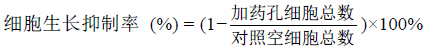
将密度为每毫升1.5×104~3×104个的MCF-7或MCF-7/Adr细胞接种于96孔板中, 每孔100 μL, 置培养箱中孵育24 h使其贴壁; 将待测化合物母液用培养液稀释成各个不同浓度后每孔加入100 μL, 与细胞共同孵育96 h; 之后每孔加入50 μL的2 mg·mL−1 MTT溶液, 置培养箱中孵育3~4 h; 甩板, 倒扣于滤纸上充分吸干残留液体后, 每孔加入200 μL DMSO于振荡器上振荡10 min以溶解蓝紫色结晶物; 使用酶标仪测570 nm处吸光值, 设A1 (含200 μL DMSO) 为空白对照孔。
由下述公式分别计算各化合物的细胞存活率:

用药物浓度的对数值与细胞存活率 (1−细胞生长抑制率) 线性回归, 求出各化合物对肿瘤细胞株的半数生长抑制浓度 (GI50)。
| [1] | Yang XL, Wang W, Tan J, et al. Synthesis of a series of novel dihydroartemisinin derivatives containing a substituted chalcone with greater cytotoxic effects in leukemia cells [J]. Bioorg Med Chem Lett, 2009, 19: 4385-4388. |
| [2] | Xie LJ, Zhai X, Ren L, et al. Design, synthesis and antitumor activity novel artemisinin derivatives using hybrid approach [J]. Chem Pharm Bull, 2011, 59: 984-990. |
| [3] | Oh S, Jeong IH, Shin WS, et al. Growth inhibition activity of thioacetal artemisinin derivatives against human umbilical vein endothelial cells [J]. Bioorg Med Chem Lett, 2003, 11: 3665-3668. |
| [4] | Jung M, Tak J, Chung WY, et al. Antiangiogenic activity of deoxoartemisinin derivatives on chorioallantoic membrane [J]. Bioorg Med Chem Lett, 2006, 16: 1227-1230. |
| [5] | Wang S, Sasaki T. Synthesis of artemisinin dimers using the Ugi reaction and their in vitro efficacy on breast cancer cells [J]. Bioorg Med Chem Lett, 2013, 23: 4424-4427. |
| [6] | Morrissey C, Gallis B, Solazzi JW, et al. Effect of artemisinin derivatives on apoptosis and cell cycle in prostate cancer cells [J]. Anticancer Drugs, 2010, 21: 423-432. |
| [7] | Efferth T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells [J]. Curr Drug Targets, 2006, 7: 407-421. |
| [8] | Wang Q, Wu LM, Zhao Y, et al. The anticancer effect of artesunate and its mechanism [J]. Acta Pharm Sin (药学学报), 2002, 37: 477-478. |
| [9] | Lu JJ, Meng LH, Cai YJ, et al. Dihydroartemisinin induces apoptosis in HL-60 leukemia cells dependent of iron and p38 mitogen-activated protein kinase activation but independent of reactive oxygen species [J]. Cancer Biol Ther, 2008, 7: 1017-1023. |
| [10] | Lai H, Singh NP. Selective cancer cell cytotoxicity from exposure to dihydroartemisinin and holotransferrin [J]. Cancer Lett, 1995, 91: 41-46. |
| [11] | Chen HH, Zhou HJ. Inhibitory effects of artesunate on angiogenesis [J]. Acta Pharm Sin (药学学报), 2004, 39: 29-33. |
| [12] | Hennessy M, Spiers JP. A primer on the mechanics of P-glycoprotein the multidrug transporter [J]. Pharm Res, 2007, 55: 1-15. |
| [13] | Yang XL, Liu D, Wang W, et al. Design, synthesis and antiproliferative activities of artemisinin derivatives containing a substituted piperazine [J]. Lett Drug Design Discov, 2009, 6: 595-598. |
| [14] | Li Y, Wu JM, Shan F, et al. Synthesis and cytotoxicity of dihydroartemisinin ethers containing cyanoarylmethyl group [J]. Bioorg Med Chem, 2003, 11: 977-984. |