 2015, Vol. 50
2015, Vol. 50

抑制HIV-1病毒的侵入和增殖以治疗艾滋病, 可通过多种环节, 例如抑制逆转录酶或蛋白酶的药物是干扰病毒的核酸复制和蛋白的成熟, 终止病毒颗粒的繁殖。另一个环节是阻断病毒向宿主细胞的侵染。
HIV侵入宿主细胞, 一个重要步骤是病毒的脂质膜与宿主的细胞膜发生融合, 启动融合的过程是通过HIV病毒包膜的表面蛋白与细胞表面的多种受体发生相互作用, 后者一个受体是CD4, 是HIV-1表面蛋白gp120结合的主要受体。但只靠CD4本身还不能足以发生融合以启动侵入, 还需要另一个共受体 (coreceptor) 参与, 这个共受体来自于趋化因子受体(chemokine receptor), 属于GPCR家族。趋化因子受体CCR5是HIV-1蛋白融合并侵入宿主细胞的主要的辅助性受体。然而, 在CCR5编码区缺失第32碱基对的纯合子证明不能感染HIV-1, 因为这些纯合子不能在细胞膜上表达有功能的CCR5受体, 以至于缺失32碱基对的人群感染HIV-1的几率与发病率很低。所以, 阻断CCR5受体的功能是治疗HIV-1病毒侵染的重要途径。
第一个抑制HIV病毒侵染的抗艾滋病药物称作恩夫韦地 (enfuvirtide), 是多肽 (36肽) 化合物, 为病毒融合蛋白的模拟物, 结合位点是gp41蛋白, 阻止病毒的侵入。恩夫韦地是Duke大学与罗氏公司共同研发、2003年FDA批准的第一个蛋白融合抑制剂, 为注射用药。
2 苗头化合物辉瑞公司研究新的抗艾滋病药物, 设定的作用环节是阻止HIV-1的gp120蛋白与宿主细胞上CCR5受体的结合, 切断HIV-1向宿主细胞的侵入。研发的目标是小分子化合物, 通过口服用药, 干扰蛋白-蛋白的相互作用, 达到治疗效果。
早期评价化合物的活性, 是测定化合物对放射性碘标记的趋化因子 (125I-MIP-1b) 与CCR5结合的竞争作用, 设定苗头化合物的标准是: IC50 < 2.5 μmol·L−1; clog P < 5; 配体效率LE > 0.2。通过对50万个化合物的大规模筛选, 发现苯并咪唑化合物1 (相对分子质量 = 409.6), 呈现弱结合活性 (IC50 = 0.65 μmol·L−1), 但没有显示抗病毒作用。此外, 1对细胞色素CYP2D6有较强抑制作用 (IC50 = 40 nmol·L−1), 应去除该不利因素。
3 苗头向先导物过渡 3.1 降低亲脂性, 消除CYP2D6的抑制作用化合物1的亲脂性较强, 在系列变换中, 二苯甲基中的一个苯基换成环丁酰胺基成为化合物2, 不仅 提高了与CCR5的结合能力, 也呈现了抑制病毒的活性, IC90 = 75 nmol·L−1 (IC90是抑制90% HIV侵染细 胞时的化合物浓度), 但仍有CYP2D6抑制作用。抑制CYP的原因可能是哌啶环与铁卟啉发生配位结合, 在哌啶环上加入基团以增加位阻, 可阻止这种结合。为此将哌啶变换成体积较大的托品环, 化合物3由于位阻和构象的限制, 避免了与CYP的结合, 而且还降低了与CCR5结合熵的损失, 两个对映体中S-异构体的抗病毒活性IC90 = 6 nmol·L−1, 高于R构型 (Amour D, de Groot M, Edwards M, et al. ChemMedChem, 2006, 1: 706−709)。然而化合物3对于人心肌钾通道hERG有较强抑制作用, 在1 μmol·L−1浓度下抑制率达到99%。这个潜在的心脏毒性须消除。
3.2 消除对hERG钾通道的抑制作用化合物3对hERG的抑制作用可能是由于分子 具有亲脂性和有碱性氮原子的缘故, 3的pKa = 7.8。基于化合物呈现抑制hERG的药效团模型 (图 1), 显 示了3的托品环上氮原子处于疏水性芳香环的中心, 推测降低化合物的碱性可消除对hERG的抑制。其 实不然。化合物4是在托品环中引入氧原子, 碱性下降到pKa = 6.0, 这样, 在生理pH条件下 (pH = 7.4), 质子化程度只有3%, 但4并没有降低对hERG的抑制 (另一个佐证是将抗心律失常药多非立特的碱性氮原子乙酰化后, 没有降低对hERG的抑制)。

|
图1 hERG钾通道抑制剂的药效团 |
另一方面, 通过对化合物3与图 1的药效团比 对, 发现环丁基相当于一个疏水性芳基, 如果在这里引入极性基团, 应降低同hERG的结合。经合成一系列化合物后, 发现化合物5仍保持抗病毒侵染活性, IC90 = 2 nmol·L−1, 消除了抑制hERG的作用, IC50 = 10 μmol·L−1 (Price DA, Armour D, De Groot M, et al. Bioorg Med Chem Lett, 2006, 16: 4633−4637)。5作为先导化合物可进一步优化。
4 结构优化 4.1 改善吸收性化合物5具有与CCR5高亲合力和抗病毒侵染活性, 也消除了抑制hERG钾通道作用。但存在药代动力学问题, 例如穿越细胞膜的性能低, 体外Caco-2实验进入细胞的速率很低 (Papp < 1×10−6 cm·s−1), 灌胃大鼠实验表明生物利用度差。原因之一可能是分子的极性表面积 (PSA) 偏大。化合物5含有7个极性原子, PSA = 76 Å2 (化合物3的PSA = 46 Å2), 氮和氧原子与水分子可形成氢键结合, 阻碍了向脂质膜的传入。为了减少极性基团, 将N-乙酰氮杂环丁烷变换成四氢吡喃, 酰胺片段换成醚键, 极性原子数减少, 化合物6的PSA降低到58 Å2, 对Caco-2细胞透入增加, Papp = 23×10−6 cm·s−1, 显著提高了大鼠的口服生物利用度, 而且保持了抗病毒活性 (IC90 = 0.6 nmol·L−1), 无hERG抑制作用。
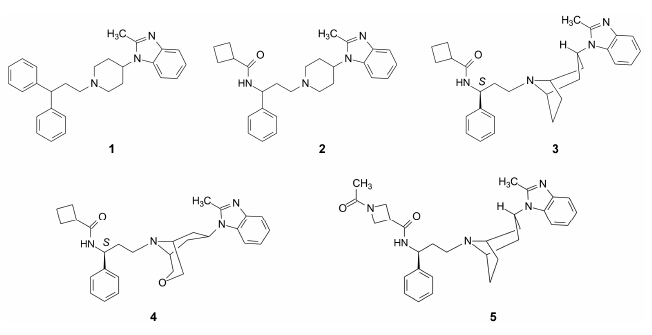
化合物6有较高的抗病毒活性, 消除了抑制hERG的作用, 也解决了过膜吸收性, 但动物体内出现易于代谢的问题。用人和犬肝细胞温孵化合物6, 清除率分别高达27和14 mL·min−1·kg−1, 犬灌胃发生广泛的首过效应, 口服生物利用度低于10%, 推测人体也会有类似现象。因此, 这条研发路径到了中止的境地。

在消除抑制hERG的作用过程中, 分子左侧的苯基和中部的哌啶环分别进行了变换, 其实, 简化苯并咪唑结构也是可行的。因为分子模拟显示, 替换或修饰苯并咪唑片段可以降低与hERG的结合。不过, 若在苯并咪唑环上引入取代基, 不仅增加了合成的难度, 也使分子尺寸加大不利于药代。因此决定用简单杂环换掉苯并咪唑环。为了探索杂环对活性、hERG结合性能和过膜性的构效关系, 用中部为哌啶片段化合物作范本化合物, 以使合成简单化。
4.3.1 苄基三唑和苄基噁二唑类化合物将并环的苯并咪唑换成苄基取代的三唑或噁二唑, 提高了与CCR5受体的结合活性 (IC50 = 5~20 nmol·L−1), 但也增强了抑制hERG作用 (IC50 = 0.1 μmol·L−1)。
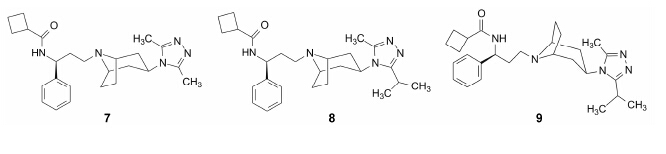
4.3.2 简单烷基取代的三唑化合物二甲基三唑化合物与CCR5的亲和力IC50 = 50 nmol·L−1, 同时抑制hERG的作用弱, 在0.1 μmol·L−1浓度下对hERG的 抑制作用5%, 提示简单取代的三唑环可将抑制CCR5的主作用与抑制hERG的不良反应分开。将二甲基 三唑连接到托品烷系列, 例如化合物7的抑制病毒 活性IC90 = 13 nmol·L−1, 在0.1 μmol·L−1浓度下抑制hERG的抑制率为10%; 将一个甲基变换为异丙基的化合物8抑制病毒的活性提高到IC90 = 8 nmol·L−1, 对hERG的影响不大, 在0.3 μmol·L−1浓度下对hERG的抑制率为30%。
4.3.3 exo-差向体优于endo-体: 里程碑式化合物化合物8的2-甲基-5-异丙基三唑环由于体积较大造成对分子构象的阻碍效应, 以致产生的两个差向异构体, exo-与endo-化合物可以分离开, exo-异构体8, 其托品环采取的是正常的椅式结构; 而endo-化合物9为了缓解三唑环与托品环桥头碳的1,3直立键的空间张力, 六元环采取了假船式构象。化合物9的抗病毒活性显著低于8, IC90 = 101 nmol·L−1。以后的优化目标保持为exo-异构体。
化合物8的分布系数logD = 1.6, 具有良好的药 代动力学性质, 犬的口服生物利用度F = 43%, 半衰期t1/2 = 7 h, 清除率Cl = 172 mL·min−1·kg−1。拟进入 开发阶段, 但在临床前全面试验对心脏的安全性中, 发现仍有延长Q-T波的潜在的心脏风险, 因而终止 了对化合物8的研究, 继续进行结构优化。
4.3.4 回到优化四元环的路径, 候选化合物马拉韦罗的确定优化至此, 分子的右端为2-甲基-5-异丙基-1, 3, 4-三唑片段, 中部为托品烷结构, S-构型的苯环也已确定。再优化的位置是环丁烷。将环丁基换成环戊基, 化合物10的logD = 2.1, 虽然亲脂性略高于化合物8, 抗病毒活性强于8, IC90 = 2 nmol·L−1, 却降低了对hERG的抑制作用, 在0.3 μmol·L−1浓度下的抑制率为18%, 推论这个位置的亲脂性与抑制hERG作用没有相关性。当用三氟丙基代替环丁基, 化合物11的logD = 1.8, 抗病毒活性IC90 = 14 nmol·L−1, 降低了对hERG的抑制作用, 在0.3 μmol·L−1浓度下的抑制率为14%。将化合物10和11的有益的结构因素 相结合, 以4, 4-二氟环己基替换环丁基, 得到的化合物12具有如下的数据: logD = 2.1, 抗病毒活性IC90 = 2 nmol·L−1, 在0.3 μmol·L−1浓度下对hERG无抑 制作用, 用膜片钳方法评价化合物12对细胞表达的hERG抑制实验, 表明在10 μmol·L−1浓度下的抑制率为19%, 说明环己基的位阻效应和二氟取代的偶极效应消除了同hERG结合。
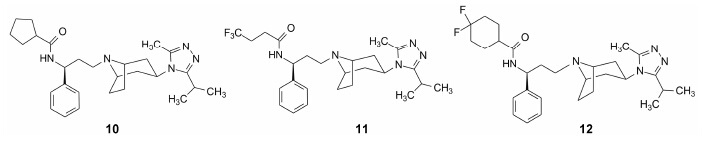
从确定苗头化合物开始, 经向先导物的过渡, 和“分段”优化活性与成药性, 最后确定了候选化合物, 总共合成了965个目标分子, 优化出的化合物12具有诸多优良的品质, 预示有良好的开发前景, 遂命名为马拉韦罗 (maraviroc), 进入临床前研究, 经三期临床试验研究, 于2007年FDA批准上市, 作为口服药治疗HIV-1感染的艾滋病 (Price DA, Armour D, De Groot M, et al. Curr Top Med Chem, 2008, 8: 1140− 1151)。
5 马拉韦罗的结合位点马拉韦罗抑制了作为辅助受体CCR5的功能, 从而阻断了HIV-1包膜蛋白与宿主细胞的表面蛋白的融合, 阻止了病毒对细胞的侵染。马拉韦罗究竟怎样同CCR5结合呢?吴蓓丽等解析了马拉韦罗与CCR5复合物的晶体结构, X射线衍射表明马拉韦罗的结合位点与趋化因子和病毒的糖蛋白gp120对CCR5的结合位点是不同的, 马拉韦罗是与CCR5的变构区结合, 导致受体构象发生改变, 非竞争性地阻止了gp120与CCR5之间的蛋白−蛋白相互作用。图 2是马拉韦罗与CCR5复合物的晶体结构 (Tan QX, Zhu Y, Li J, et al. Science, 2013, 341: 1387−1390)。在药物化学和构效关系指引下, 成功地研发出抑制病毒侵入环节的第一个口服小分子药物马拉韦罗, 又从微观上诠释了结合特征和可能的作用机制。

|
图2 马拉韦罗与CCR5复合物的晶体结构 |