 2015, Vol. 50
2015, Vol. 50



哺乳动物内源性雌激素在维持机体生长、发育、繁殖等多个生命过程中起着重要作用。雌激素主要 通过细胞核中的核转录因子雌激素受体 (estrogen receptors,ERs) 起作用。ERs属于核受体(nuclear receptors) 超家族,目前已发现有两种亚型,即ERα和ERβ[1]。二者在许多组织和细胞中都有表达,但在不同组织细胞的表达程度不同,且具有不同的生理功能。ERα主要存在于子宫、卵巢 (内膜细胞)、乳腺、骨组织、肝脏以及脂肪等组织器官,在调节女性的生殖功能、代谢调控、维持骨密度等方面起重要作用。ERβ则主要分布于泌尿生殖器 (前列腺上皮细胞和膀胱)、结肠、中枢神经系统以及免疫系统等,在乳腺、前列腺和结肠细胞中有抗细胞增殖的作用。此外,ERα和ERβ在维持卵巢功能和保护心血管系统方面都起着重要作用[2,3,4]。
ERα和ERβ通过招募不同的辅因子结合到DNA的不同位点,来调控不同基因的表达。研究发现激活ERα可能导致乳腺及子宫细胞的增生,增加乳腺癌或子宫内膜癌的发病风险,而选择性激动ERβ则不会引起相应的细胞增殖作用,反而有一定的抗细胞增殖作用。这表明具有ERβ选择性的调节剂不仅能发挥类似荷尔蒙替代疗法 (hormone replacement therapy,HRT) 中药物的治疗作用,而且能有效避免HRT治疗中产生的副作用。因此ERβ已成为研发治疗多种疾病药物的一个非常有潜力的靶标,如癌症 (乳腺癌、结肠癌、前列腺癌、间皮瘤)、良性前列腺增生症、心血管疾病、多发性硬化症、类风湿关节炎、代谢病变、炎症性肠病以及阿尔茨海默病等[5, 6]。
近年来,寻找ERβ选择性配体逐渐成为研究热点,但由于ERα和ERβ在结构上的高度相似性 (在配体结合口袋的序列同一性甚至达到了97%),设计和研究有亚型选择性的配体仍是一项重大的挑战。本文主要对近年来报道的具有选择性的ERβ调节剂进行分类介绍,并作了简单的构效关系分析。
1 ERβ选择性激动剂目前已报道的ERβ激动剂的结构具有多样性,按照母核的结构类型可以分为甾体类以及非甾体类化合物两大类。由于甾体类化合物与天然底物雌激素结构类似,在此主要介绍有潜在临床应用的甾体类化合物,以及结构新颖的非甾体类选择性激动剂。
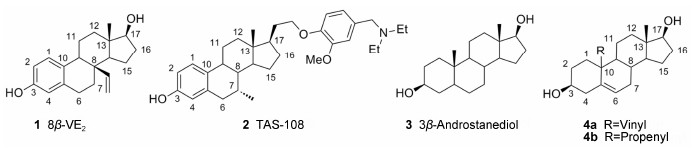
1.1 甾体类ERβ激动剂 1.1.1 雌二醇衍生物雌二醇 (E2) 是活性最高的ER内源性配体。2004年Hillisch等[7]报道了以E2 为起始结构设计的ERβ选择性配体。在E2的8β位引入乙烯基,得到的8β-VE2 (化合物1) 是ERβ选择性激动剂。与E2 [EC50 (ERα) = 0.006 1 nmol·L−1,EC50 (ERβ) = 0.023nmol·L−1] 相比,8β-VE2对ERα的细胞转录活性降低 (EC50 = 2.3nmol·L−1),而对ERβ活性保持不变 (EC50 = 0.050 nmol·L−1),因而极大地提高了ERβ的选择性 (β/α = 46)。
2005年Yamamoto等[8]对E2进行进一步结构修饰,7α位用甲基取代,同时17β-OH用连有取代基的芳基取代,得到化合物2 (TAS-108)。TAS-108对ERs结合亲和力 [IC50 (ERα) = 11 nmol·L−1,IC50(ERβ) = 5.6 nmol·L−1,IC50值由竞争性的放射或荧光配体结合实验测得] 与E2相当[IC50 (ERα) = 0.2nmol·L−1,IC50(ERβ) = 0.5 nmol·L−1],虽然其对ERα和ERβ的结合亲和力差别不大,但其在细胞实验中表现为ERα的完全拮抗活性,而对ERβ则是转录激动活性。在这之前报道的ER配体中都没有类似的作用,因此化合物TAS-108有很大的临床应用意义。2012年Inaji等[9]报道了TAS-108的随机II期临床试验结果,TAS-108在每天40、80和120 mg的剂量下都没有表现出明显的不良反应。因此TAS-108有望成为HRT治疗中新的二线或三线药物。
研究发现某些甾体激素的代谢产物也具有ERβ选择性[10],如3β-二氢雄甾烷二醇 (化合物3)。化合物3的结合亲和力较高 [ERβ-RBA = 7%,ERα-RBA = 3%,RBA(relative-binding affinity) 表示以E2 (100%) 为参考的相对结合亲和力值,由结合亲和力实验中测得的IC50值计算得到],但ER亚型选择性较弱。化合物3在细胞实验中不但有较高的ERβ激动活性 (EC50 = 23 nmol·L−1),其EC50值与E2的代谢产物雌 三醇 (EC50 = 17nmol·L−1) 相当,而且ERβ选择性较雌三醇高。2014年,Piccolella和Colciago等[11, 12]分别报道了化合物3有抑制前列腺细胞增殖和转移的作用。
1.1.2 雄甾烯衍生物Merck公司研究人员发现10位乙烯基取代的雄甾烯衍生物具有较高的ERβ选择性[13]。如化合物4a在结合亲和力实验中对ERβ的 (IC50为11 nmol·L−1) 选择性是ERα的212倍; 在 细胞实验中同样对ERβ表现出较高的活性 (EC50为 4 nmol·L−1),选择性高达246倍。但是遗憾的是,化合物4a也是雄激素受体 (AR) 激动剂 (IC50为33 nmol·L−1),即在ER/AR之间的选择性较差。将化合物4a的10位用丙烯基取代 (化合物4b),则在ERβ与AR之间的选择性增加,达到45倍,同时在结合亲和力实验中也保持了较高的ERβ选择性(β/α = 126)。在细胞实验中,虽然4b对ERβ仍有较强激活作用 (EC50 = 21 nmol·L−1),但对ERβ的选择性降低 (β/α = 16)。
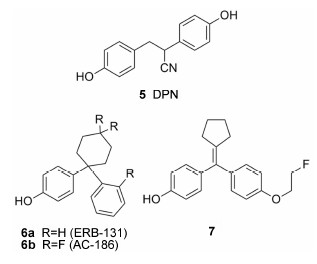
自小分子化合物DPN (化合物5) (β/α = 72,ERβ-EC50为0.66 nmol·L−1)被报道作为ERβ选择性激动剂始,即被作为参考分子用于ERβ选择性配体的研究[14]。2012年Carroll等[15]报道了DPN分子的两种对映异构体的合成及活性差异。实验表明两种异构体均有较高的亲和力和选择性,其中R-构型 (RBA-ERβ = 32.6%,RBA-ERα = 0.098%,β/α = 332) 较S-构型 (RBA-ERβ = 9.7%,RBA-ERα = 0.066%,β/α = 147)有更高的结合亲和力和选择性。因此在今后的ERβ生物功能研究实验中,R-DPN分子将是更好的选择。
2009年Piu等[16, 17]报道了二芳基环己烷衍生 物具有ERβ选择性,其中化合物6a (ERB-131) 对 ERβ选择性大于100倍,且EC50值达到纳摩尔级别。ERB-131作为ERβ选择性激动剂在其他核受体中并不表现出激动作用,在体内实验中对ERα也没有作用。此外,ERB-131在炎症模型中表现出较好的抑制作用,可用于炎症性疼痛的治疗。在二芳基环己烷的骨架上进行进一步的结构改造,引入3个氟原子得到化合物6b (AC-186),选择性达到800倍以上 (EC50-ERβ为6 nmol·L−1,EC50-ERα为5 000 nmol·L−1)。AC-186在阿尔茨海默病和帕金森病小鼠模型中有体内激动活性。
Zhu等[18]于2012年报道了二芳基环戊烷衍生物同样有ER活性,其中化合物7对ERβ有较好的选择性 (β/α = 6.4),其结合亲和力与雌二醇相当 (RBA = 88.6%);对ERα的结合亲和力较弱,RBA值仅有13.8%。18F原子标记的生物分布实验显示,化合物7在ER阳性组织细胞如子宫、卵巢、肾脏及脾脏被选择性吸收。由于亲脂性的小分子化合物容易通过血脑屏障,在脑中有一定的积累,因此化合物7有望成为医学诊断成像中ERs的探针化合物,用于测定乳腺癌细胞中ERβ的含量。
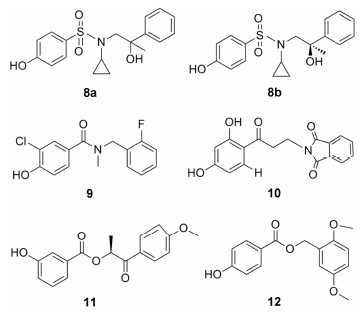
2011年Roberts等[19]报道了羟基苯磺酰类化合物对ERβ具有很高 的选择性,其中化合物8a表现出较高的活性及选 择性 (EC50-ERβ为101nmol·L−1,EC50-ERα为2 310nmol·L−1)。将化合物8a进行手性拆分,得到其中一个异构体8b具有更好的药理学性质,不仅选择性大大增加,对ERβ的活性也有所提高 (EC50-ERβ为79 nmol·L−1,EC50-ERα > 5 400nmol·L−1)。化合物8b与ERβ复合物的晶体结构显示 (图 1),叔羟基与磺酰基的氧原子形成分子内氢键,导致苯基朝向His475残基,同时环丙基与Ile373残基形成疏水相互作用。磺酰胺基的氧原子与Met336形成范德华相互作用。而在ERα结构中,ERβ中的Ile373与Met336分别被Met421和Leu384取代,从而导致了化合物8b与ERα的结合力减弱。

|
图 1 ERβ-8b的复合物晶体结构 (PDB号: 2YLY),图中原子间距离的单位为0.1 nm |
本课题组Chen等[20]最近报道了利用基于配体的药效团匹配和基于受体的虚拟筛选方法,从Maybridge和Enamine数据库筛选得到一系列EC50值小于10 µmol·L−1的ERβ激动剂 (化合物9~12),其酵母双杂交 (Y2H) 实验结果如表 1所示。这些化合物在分子水平及细胞水平上对ERα都没有明显的活性,表明对ERβ具有很好的选择性。
|
|
Table 1 化合物9~12对ERβ的激动活性. REC10(relative effective concentration): 达到E2激活效应的10%时化合物的浓度 |
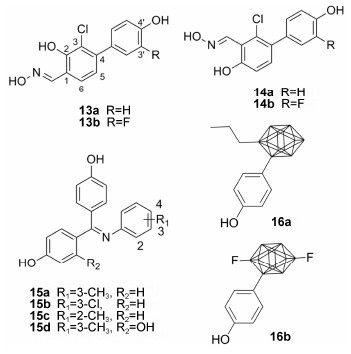
2008年Minutolo等[21]报道了单芳基取代的水杨醛肟类化合物的ERβ选择性作用。在这一系列化合物中,3-C位氯原子取代的化合物13a选择性和结合亲和力最高 (ERβ-RBA = 4.21%,ERα-RBA = 0.065%,β/α = 65)。在细胞实验中,化合 物13a虽然表现出ERβ的部分激动作用 (EC50为11 nmol·L−1),但是相比于结合亲和力,ERβ选择性减弱 (EC50-ERα = 26nmol·L−1),因此需要对其进行进一步的结构修饰。
该课题组[22]于2009年报道了基于化合物13a的衍生物。其中,同时在3-C位引入氯原子和3'-C位引入氟原子的化合物13b结合力 (Ki为7.1 nmol·L−1) 和选择性 (β/α = 62) 最好。更重要的是,化合物13b在细胞实验中也表现出ERβ选择性的完全激动活性,EC50值达到4.8 nmol·L−1,且选择性比化合物13a增加 (EC50-ERα为19 nmol·L−1)。
2011年该课题组对水杨醛肟类化合物进行进一步的结构改造[23]。化合物13a和化合物13b的羟基与肟基的位置互换得到的化合物14a和14b,结合亲和力进一步提高,Ki值分别达到了0.38 nmol·L−1和0.57 nmol·L−1,同时两个化合物均保持了较高的ERβ选择性 (β/α分别为29倍和46倍)。在细胞水平实验中,化合物14a (EC50为0.23 nmol·L−1) 和14b (EC50为1.3 nmol·L−1)对ERβ同样表现出完全激动作用,且化合物14b相比于内源性的雌二醇表现出50倍以上的选择性。
Liao等[24]在2014年报道了三芳基取代的亚胺 类化合物对ERβ有较高的结合亲和力。其中3-甲基取代的化合物15a选择性最高 (β/α = 5.2,ERβ-RBA = 69.2%); 用氯原子取代的化合物15b (β/α = 2.5,ERβ-RBA = 114%) 以及2-甲基取代的化合物15c (β/α = 2.8,ERβ-RBA = 116%) 对ERβ结合亲和力增加,但是选择性降低。若在对羟基苯酚取代基上再引入OH,则选择性和结合亲和力均下降,如化合物15d (β/α = 2.5,ERβ-RBA = 15.3%)。
2014年Ohta等[25]报道了碳硼烷类化合物能选择性作用于ERβ,设计并合成了一系列对羟基苯酚取代化合物。实验证明在碳硼烷的邻位引入长脂肪链取代基比在间位引入取代基选择性高,如化合物16a的选择性最高为7.4倍。在结合亲和力实验中与ERβ结合的RBA值为87% (ERα-RBA = 11.8%)。该课题组[26]在2013年报道了两个氟原子取代的碳硼烷化合物同样具有ERβ选择性,如化合物16b (β/α = 10.1,ERβ-RBA= 49.9%)。改变氟原子的位置或者用其他的取代基替换,则选择性降低。
1.2.5 芳香单环类化合物 (噻吩衍生物)Min等[27]于2013年报道了一类以噻吩环为母核的高选择性的ERβ配体。其中化合物17a的选择性最高 (β/α = 16),其结合亲和力ERβ-RBA为33.1%。实验表明,苯酚环上的羟基位置对活性影响较大,间羟基苯酚衍生物比对羟基苯酚衍生物的结合亲和力有明显降低,如化合物18 (β/α = 8.3,ERβ-RBA = 0.066%)。另外,苯酚环上的取代基同样对活性有一定的影响,如氯原子取代的化合物17b选择性和结合力均下降 (β/α = 1.5,ERβ-RBA = 10.0%)。该课题组用呋喃环替代噻吩环合成了一系列化合物并进行生物测试,总体上呋喃环为母核的化合物较噻吩类化合物结合亲和力减弱。

苯并吡喃衍生物 2006年Norman等[28]以ERβ选择性激动剂19a (β/α = 8) 为参考分子,分别得到其两个对映异构体SERBA-1 (化合物19b) 和SERBA-2 (19c)。SERBA-1(EC50为0.66 nmol·L−1,β/α = 32) 的选择性和结合力比SERBA-2略高。2007年Richardson等[29]报道了氟取代的化合物LY3201 (20) 同样具有ERβ选择性,其与ERβ结合的Ki值达到0.44nmol·L−1,活性是ERα的19倍。2012年Suzuki等[30]报道了LY3201对中枢神经系统疾病有潜在的治疗作用。
2012年Shi等[31]报道了7-OH修饰的genistein衍生物GS-14 (21) 具有多靶标作用,即对乙酰胆碱酯酶的抑制作用、雌激素受体的激活作用以及中枢神经系统的保护作用。MCF-7细胞增殖实验结果显示,GS-14的ERβ激动效应与genistein相当,同时分子对接模拟结果的结合自由能显示GS-14对ERβ具有较高的选择性。该化合物有望成为治疗阿尔茨海默症的多靶标化合物。

本课题组Shen等[32]利用基于受体的虚拟筛选方法,从SPECS分子库中筛选出18个有效的ER配 体,其中有5个化合物 (22~24) 的EC50值低于100 nmol·L−1 (酵母双杂交实验测试结果如表 2)。化合物22a对ERα和ERβ都表现出较高的激动活性。若将R1的氯原子用甲基取代 (22b),则对ERα活性增加,对ERβ活性减弱,选择性降低。在R2位引入氟原子 (22c),对ERα和β的活性均减弱,但是选择性有所提高。在苯并吡喃母核与苯环取代基之间插入极性氧原子(23),则对ERα的活性显著降低,即选择性提高。将苯环取代基并入苯并吡喃母核中的三元稠环化合物24,对ERβ活性变化不大,对ERα作用大大降低,选择性达到近30倍。

|
|
Table 2 化合物22~24对ERα和ERβ的激动活性 |
萘衍生物 Mewshaw等[33]报道了1-苯基萘化合物25a (IC50为5 nmol·L−1,β/α = 39),对ERβ的结合 力和选择性较高。在苯环上引入氟原子得到的化合物25b,结合亲和力进一步增加 (IC50为3.8 nmol·L−1,β/α = 49)。研究发现取代基位置对活性影响较大,如C8-位氰基取代的化合物26a (IC50为2.7 nmol·L−1,β/α = 78) 对ERβ的选择性明显高于C4-位氰基取代的化合物26b (IC50为2 nmol·L−1,β/α = 43)。
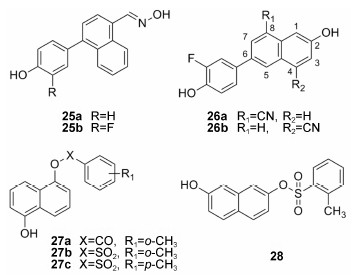
2012年本课题组Shen等[34]通过组合基于配体和基于受体的虚拟筛选方法,发现一系列萘类化合物具有ERβ选择性作用。其中化合物27、28在酵母双杂交试验中,对ERβ表现为激动作用,对ERα表现为拮抗作用。这类化合物被称作为双效化合物,不仅在临床方面有较大的应用价值,而且可以作为分子探针研究ER亚型之间的差别,进一步阐明不同亚型与不同疾病之间的作用机制。对比化合物27a (ERβ- EC50 = 2.69μmol·L−1,ERα-IC50 = 0.43μmol·L−1)、27b (ERβ-EC50 = 0.229 μmol·L−1,ERα-IC50 = 0.255 μmol·L−1)、27c (ERβ-EC50 = 0.84 μmol·L−1,ERα-IC50 = 2.61μmol·L−1) 和28 (ERβ-EC50 = 0.094μmol·L−1,ERα- IC50 = 8.77 μmol·L−1) 可以看出,将化合物羰基替换成磺酰基,则对ERβ的结合力大大提高,说明磺酰基是较羰基更好的链接基团。另外,化合物27c在MCF-7细胞实验中表现出很高的抗MCF-7细胞增殖能力,细胞抑制率达到74.74%。化合物28在细胞实验中表现出较高的ERβ激动活性 (EC50为3.55 μmol·L−1),其最大效应值甚至超过了内源性配体雌二醇。
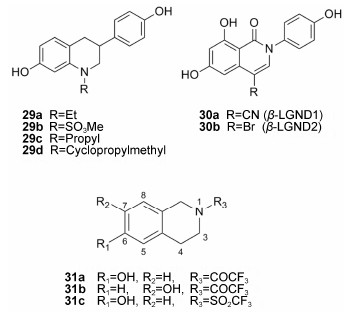
喹啉和喹唑啉酮衍生物 Chen等[35]于2007年报道了四氢喹啉类化合物具有ERβ选择性。乙基取代的化合物29a对ERβ结合亲和力最高,IC50值达到 83 nmol·L−1,但是选择性较低 (β/α = 17)。甲磺酰基取代的化合物29b选择性提高 (β/α = 61),但是对ERβ的结合力减弱 (IC50为520 nmol·L−1)。值得一提的是,丙基取代的化合物29c (EC50-ERβ为4.75 μmol·L−1,IC50-ERα为2.65 μmol·L−1) 和甲基环丙基取代的化合物29d (EC50-ERβ为6.89μmol·L−1,IC50-ERα为5.57μmol·L−1) 在酵母双杂交实验中表现出ERβ激动活性和ERα拮抗作用,这与之前提到的化合物TAS-108类似,具有一定的临床应用意义。
由GTx公司研发的异喹啉酮类化合物β-LGND1 (30a) 和β-LGND2 (30b)是选择性的ERβ激动剂[36],其ERβ选择性大于100倍,且与其他核受体没有作用。在诱导肥胖的动物模型中,能有效地预防和治疗肥胖。2012年Giddabasappa等[37]报道了β-LGND2有促进ERβ介导的体内和体外抗血管生成作用,可用于由血管生成失调引起的糖尿病性视网膜病和其他类似的眼部疾病。2013年Pedram等[38]报道了β-LGND2在心血管疾病方面的应用。
2011年Möcklinghoff等[39]通过基于片段的筛选方法,发现四羟基喹啉类片段小分子有ERβ选择性活性。其中化合物31a对ERβ有较高的活性和选择性。若将其6-OH换成7-OH (31b),则对ERβ的选择性和活性均下降。将羰基用磺酰基取代得到的化合物31c选择性与活性则没有较大的变化。有趣的是,晶体结构显示(图 2),这3个化合物在受体口袋内处于相同的位置,化合物31a与31c的羟基能与Glu305、Arg346以及口袋内的一个水分子形成经典的氢键网络,而化合物31b的7-OH则偏离羟基网络的中心位置,仅能与Glu305形成一个氢键作用。这样的结合模式也恰好解释了化合物31b与受体作用活性减弱的原因。分析化合物31c在ERβ中的结合模式发现,其磺酰基接近于Met421残基,而在ERα活性口袋中相应的位置,其硫原子以及甲基上的氟原子则可能与体积较大的Ile373残基形成碰撞,这也可能是化合物31c对ERβ的结合作用更强的原因。结合以上分析,这些片段小分子化合物可以作为起始结构进一步设计选择性更高的配体化合物。

|
图 2 (A) ERβ-31a、ERβ-31c的复合物晶体结构 (PDB号: 3OMO、3OMQ); (B) ERβ-31b的复合物晶体结构 (PDB号: 3OMP); 图中原子间距离的单位为0.1 nm |
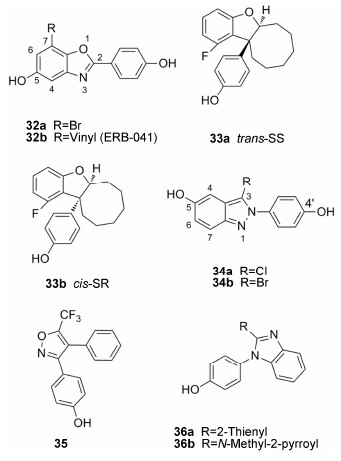
苯并噁唑、苯并呋喃衍生物 Manas等[40]报道 了一系列的苯并噁唑类ERβ选择性化合物。如7-溴代化合物32a在HEK293细胞转录实验中的EC50 值达到了纳摩尔级别,而且β/α选择性达到363倍。7-乙烯基取代的化合物ERB-041 (32b)显示出200倍的ERβ选择性和很好的结合亲和力 (IC50为5.4 nmol·L−1)。最近,ERB-041作为治疗子宫内膜异位症的药物进入临床II期试验评价阶段[41],但是结果尚未公布。
2013年,Sundén等[42]报道了二羟基苯并呋喃类化合物具有ERβ选择性激动作用。其中化合物33a活性最强,其EC50值小于1 nmol·L−1,通过不对称合成得到的其对映异构体33b,选择性更是达到了1 000倍以上,而且对ERβ同样有较强的激动效应 (EC50为10 nmol·L−1)。值得一提的是,化合物33a在接近10 μmol·L−1浓度时,对ERα也仅表现出部分激动效应。
吲哚、吲唑、苯并咪唑衍生物 De Angelis等[43]报道了以苯基吲唑为母核的结构衍生物有较高的ERβ选择性,尤其是在C-3位有极性基团的化合物有的选择性更高,如化合物34a和34b的选择性分别达到107倍 (ERβ-RBA = 32%,ERα-RBA= 0.3%) 和102倍 (ERβ-RBA = 18.2%,ERα-RBA= 0.18%)。它们在细胞转录实验中同样对ERβ表现出选择性的激动作用。对这一系列化合物的活性测试结果显示,4'-OH以及C-3的极性取代基是决定化合物选择性和结合力的关键,去掉4'-OH则化合物几乎没有结合作用 (RBA- ERs < 0.004),C-3位无取代或用非极性基团如甲基、乙基、丙基等取代时,选择性和结合亲和力都有不同程度的下降。
Chesworth等[44]报道了以不同杂环为母核的ERβ选择性配体,其中化合物35对ERβ的结合亲和力 (IC50为2.4 nmol·L−1) 较高,但选择性一般 (β/α = 3.5)。为了提高化合物35的ERβ选择性,作者合成了一系列吲哚衍生物。化合物36a (IC50-ERβ为49.7 nmol·L−1) 和36b (IC50-ERβ为28.1nmol·L−1) 虽然对ERβ的结合亲和力有所降低,但是选择性分别达到了126和105倍。在细胞水平实验中,化合物36b对ERβ表现为完全激动活性,而对ERα只有微弱的激活作用,因此,化合物36b在细胞水平同样具有较高的选择性。
尽管与选择性ERβ拮抗剂相关的治疗作用还未有报道,但是具有高活性的ERβ选择性拮抗剂仍是研究ERβ生理功能的重要工具。到目前为止,关于ERβ选择性拮抗剂的报道还很有限。
2.1 甾体类衍生物Shiau等[45]报道的化合物37 (THC) 是一个在生物效应上表现亚型选择性的化合物,对ERβ是完全拮抗作用 (IC50 < 15 nmol·L−1),对ERα则是完全激动作用 (EC50约为3 nmol·L−1)。THC与ERs的复合物晶体结构显示,在ERα中螺旋12处于激活构象的位置,利于共激活因子的结合,发挥激动效应; 而在ERβ中螺旋12能稳定存在于阻碍共激活因子与受体的结合的位置,也就是发挥拮抗作用。但是对比THC与ERs的拮抗剂 (如raloxifene) 的结构发现,THC并没有长链结构来将螺旋12推向拮抗构象,而是通过与配体结合口袋内的关键残基的作用来稳定螺旋12的位置,这种拮抗的方式可以称作为“被动拮抗”,也是雌激素受体中一种新的拮抗剂的结合模式。因此THC与ERα和ERβ的配体结合区的晶体复合物结构是研究ERα和ERβ选择性配体的非常有价值的工具。
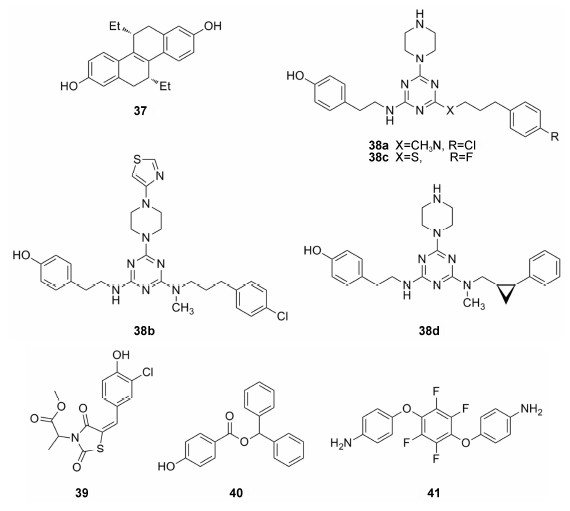
Henke等[46]报道了1,3,5-三嗪类ERβ选择性拮抗剂。在结合亲和力实验中化合物38a (IC50为15 nmol·L−1) 和化合物38b (IC50为25 nmol·L−1) 分别有25倍和30倍的选择性,且在细胞实验中对ERβ表现为完全拮抗活性,对ERα为微弱的部分激动作用。化合物38c (IC50为15 nmol·L−1,β/α = 10) 与ERβ的复合物结构显示,对氯苯基朝向Ile373残基,而在ERα相应的位置由于体积较大的Met421替代了Ile373残基,产生了较大的空间位阻作用,可能不利于化合物的结合,这样的结合模式可能是导致化合物ERβ选择性的原因。根据晶体结构中的结合模式改造得到的化合物38d选择性有所提高 (IC50为40 nmol·L−1,β/α = 31)。
2.3 其他类化合物本课题组通过几轮虚拟筛选的方法[20, 32, 34],从SPECS等商业数据库中筛选得到了部分骨架结构新颖、活性较高的ERβ拮抗剂,如化合物39~41的IC50值均小于1 µmol·L−1,其中化合物39 (IC50-ERβ为0.049 7 µmol·L−1,IC50-ERα为2.32 µmol·L−1) 的ERβ选择性最高,达到了46.7倍。但遗憾的是化合物40、41的选择性较低,对ERα也表现为拮抗作用,因此为了提高化合物的选择性,仍需要进一步的结构修饰。
3 展望从临床角度看,ERβ选择性激动剂可用于癌症、炎性以及绝经后妇女的更年期症状等疾病的治疗。由于激活ERα受体能导致细胞增殖等不良反应,所以发现新型靶向ERβ的选择性配体有重要的意义。尽管目前已有多种有亚型选择性的ERβ配体被报道,部分化合物的研发也在不断发展,但发现兼具活性、选择性以及良好药代性质的配体还存在一定的挑战。
从受体结构的角度来说,ERα和ERβ的配体结合区有59%的同一性 (sequence identity),而且在配体结合口袋附近仅有两对氨基酸残基不同,即ERα的Leu384和Met421残基,在ERβ中分别被Met336与Ile373残基替代。从已有的ER晶体结构分析,ERβ的配体结合口袋比ERα略窄。所以仅仅依靠这种细微的差别,从化合物分子与受体的结合模式出发来设计具有亚型选择性的配体分子仍面临着巨大的挑战。因此总结已有的亚型选择性的配体分子的构效关系,对ERα和ERβ的选择性配体的结构特点进行比较分析,并尝试与受体结构结合,找出除配体结合口袋以外的与选择性相关的其他区域,从相互作用的机制角度出发寻找并解释亚型选择性的原因,将是提高选择性ERβ配体设计成功率的有效途径。
从配体的角度来说,对已有的ERs配体与受体的结合模式分析发现,绝大多数具有高结合亲和力的ERs配体都具有一个保守的羟基取代基,且这一个羟基与受体结构中的Glu305残基和Arg346残基以及一个水分子形成保守的氢键网络,对与受体分子的结合起到关键作用。但是后续实验发现,由于羟基在体内易被葡萄糖醛酸代谢,因此通常化合物的药代动力学性质较差 (如口服生物利用度低、半衰期短等),这给它们的临床应用造成了一定的限制。为了提高化合物的药代动力学性质,一方面,可以利用电子等排原理,在羟基附近引入吸电子基或者具有空间位阻的基团,既能保持化合物分子的结合亲和力又能改善其药代动力学性质; 另一方面,利用前药设计原理,将羟基基团隐藏或者衍生化,也能提高化合物的药代性质。综上所述,在药物设计初期,考虑化合物的药代动力学性质,避免高活性及高选择性的化合物在临床试验阶段被淘汰,是对ER亚型选择性配体设计的另一个重要要求。
| [1] | Brzozowski AM, Pike ACW, Dauter Z, et al. Molecular basis of agonism and antagonism in the oestrogen receptor [J]. Nature, 1997, 387: 753-758. |
| [2] | Jordan VC. Antiestrogens and selective estrogen receptor modulators as multi-functional medicines 1. Receptor interacttions [J]. J Med Chem, 2003, 46: 883-908. |
| [3] | Jordan VC. Antiestrogens and selective estrogen receptor modulators as multi-functional medicines 2. Clinical considerations and new agents [J]. J Med Chem, 2003, 46: 1081-1111. |
| [4] | Luo L, Zhao SJ, Wang ZT, et al. Expression of human estrogen receptor α and β in Escherichia coli [J]. Acta Pharm Sin (药学学报), 2012, 47: 1399-1402. |
| [5] | Nilsson S, Koehler KF, Gustafsson JÅ. Development of subtype-selective oestrogen receptor-based therapeutics [J]. Nat Rev Drug Discov, 2011, 10: 778-792. |
| [6] | Liu RT, Lü QJ. Progress in the research on multi-target-directed drugs against Alzheimer's disease [J]. Acta Pharm Sin (药学学报), 2009, 44: 258-263. |
| [7] | Hillisch A, Peters O, Kosemund D, et al. Dissecting physiological roles of estrogen receptor α and β with potent selective ligands from structure-based design [J]. Mol Endocrinol, 2004, 18: 1599-1609. |
| [8] | Yamamoto Y, Shibata J, Yonekura K, et al. TAS-108, a novel oral steroidal antiestrogenic agent, is a pure antagonist on estrogen receptor α and a partial agonist on estrogen receptor β with low uterotrophic effect [J]. Clin Cancer Res, 2005, 11: 315-322. |
| [9] | Inaji H, Iwata H, Nakayama T, et al. Randomized phase II study of three doses of oral TAS-108 in postmenopausal patients with metastatic breast cancer [J]. Cancer Sci, 2012, 103: 1708-1713. |
| [10] | Kuiper GGJM, Shughrue PJ, Merchenthaler I, et al. The estrogen receptor β subtype: a novel mediator of estrogen action in neuroendocrine systems [J]. Front Neuroendocrinol, 1998, 19: 253-286. |
| [11] | Piccolella M, Crippa V, Messi V, et al. Modulators of estrogen receptor inhibit proliferation and migration of prostate cancer cells [J]. Pharmacol Res, 2014, 79: 13-20. |
| [12] | Colciago A, Ruscica M, Mornati O, et al. In vitro chronic administration of ERβ selective ligands and prostate cancer cell growth: hypotheses on the selective role of 3beta-adiol in AR-positive RV1 cells [J]. Biomed Res Int, 2014, 2014: 801473. |
| [13] | Blizzard TA. Selective estrogen receptor modulator medicinal chemistry at Merck. A review [J]. Curr Top Med Chem, 2008, 8: 792-812. |
| [14] | Meyers MJ, Sun J, Carlson KE, et al. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues [J]. J Med Chem, 2001, 44: 4230-4251. |
| [15] | Carroll VM, Jeyakumar M, Carlson KE, et al. Diarylpropionitrile (DPN) enantiomers: synthesis and evaluation of estrogen receptor β-selective ligands [J]. J Med Chem, 2012, 55: 528-537. |
| [16] | Piu F, Cheevers C, Hyldtoft L, et al. Broad modulation of neuropathic pain states by a selective estrogen receptor beta agonist [J]. Eur J Pharmacol, 2009, 590: 423-429. |
| [17] | McFarland K, Price DL, Davis CN, et al. AC-186, a selective nonsteroidal estrogen receptor β agonist, shows gender specific neuroprotection in a Parkinson's disease rat model [J]. ACS Chem Neurosci, 2013, 4: 1249-1255. |
| [18] | Zhu H, Yang Z, Lin JG, et al. Synthesis and evaluation of fluoroethyl cyclofenil analogs: models for potential estrogen receptor imaging agent [J]. J Fluorine Chem, 2012, 139: 46-52. |
| [19] | Roberts LR, Armor D, Barker C, et al. Sulfonamides as selective oestrogen receptor β agonists [J]. Bioorg Med Chem Lett, 2011, 21: 5680-5683. |
| [20] | Chen L, Wu D, Bian HP, et al. Discovery of highly selective ligands of estrogen receptor β by pharmacophore mapping and structure-based virtual screening [J]. Acta Pharmacol Sin, 2014, 35: 1333-1341. |
| [21] | Minutolo F, Bellini R, Bertini S, et al. Monoaryl-substituted salicylaldoximes as ligands for estrogen receptor β [J]. J Med Chem, 2008, 51: 1344-1351. |
| [22] | Minutolo F, Bertini S, Granchi C, et al. Structural evolutions of salicylaldoximes as selective agonists for estrogen receptor β [J]. J Med Chem, 2009, 52: 858-867. |
| [23] | Bertini S, De Cupertinis A, Granchi C, et al. Selective and potent agonists for estrogen receptor beta derived from molecular refinements of salicylaldoximes [J]. Eur J Med Chem, 2011, 46: 2453-2462. |
| [24] | Liao ZQ, Dong C, Carlson KE, et al. Triaryl-substituted Schiff-bases are high-affinity subtype-selective ligands for the estrogen receptor [J]. J Med Chem, 2014, 57: 3532-3545. |
| [25] | Ohta K, Ogawa T, Kaise A, et al. Aliphatic substitution of O-carboranyl phenols enhances estrogen receptor beta selectivity [J]. Chem Pharm Bull, 2014, 62: 386-391. |
| [26] | Ohta K, Ogawa T, Kaise A, et al. Enhanced estrogen receptor beta (ERβ) selectivity of fluorinated carborane-containing ER modulators [J]. Bioorg Med Chem Lett, 2013, 23: 6555-6558. |
| [27] | Min J, Wang P, Srinivasan C, et al. Thiophene-core estrogen receptor ligands having superagonist activity [J]. J Med Chem, 2013, 56: 3346-3366. |
| [28] | Norman BH, Dodge JA, Richardson TI, et al. Benzopyrans are selective estrogen receptor β agonists with novel activity in models of benign prostatic hyperplasia [J]. J Med Chem, 2006, 49: 6155-6157. |
| [29] | Richardson TI, Norman BH, Lugar CW, et al. Benzopyrans as selective estrogen receptor β agonists (SERBAs). Part 2: Structure-activity relationship studies on the benzopyran scaffold [J]. Bioorg Med Chem Lett, 2007, 17: 3570-3574. |
| [30] | Suzuki H, Barros RPA, Sugiyama N, et al. Involvement of estrogen receptor β in maintenance of serotonergic neurons of the dorsal raphe[J]. Mol Psychiatry, 2012, 18: 674-680. |
| [31] | Shi DH, Yan ZQ, Zhang LN, et al. A novel 7-O-modified genistein derivative with acetylcholinesterase inhibitory effect, estrogenic activity and neuroprotective effect [J]. Arch Pharm Res, 2012, 35: 1645-1654. |
| [32] | Shen J, Tan C, Zhang Y, et al. Discovery of potent ligands for estrogen receptor β by structure-based virtual screening [J]. J Med Chem, 2010, 53: 5361-5365. |
| [33] | Mewshaw RE, Manas ES, Harris HA, et al. ERβ ligands. Part 5: synthesis and structure-activity relationships of a series of 4-hydroxyphenyl-aryl-carbaldehyde oxime derivatives [J]. Bioorg Med Chem Lett, 2007, 17: 902-906. |
| [34] | Shen J, Jiang J, Kuang G, et al. Discovery and structure-activity analysis of selective estrogen receptor modulators via similarity-based virtual screening [J]. Eur J Med Chem, 2012, 54: 188-196. |
| [35] | Chen W, Lin Z, Ning M, et al. Aza analogues of equol: novel ligands for estrogen receptor β [J]. Bioorg Med Chem, 2007, 15: 5828-5836. |
| [36] | Yepuru M, Eswaraka J, Kearbey JD, et al. Estrogen receptor-β-selective ligands alleviate high-fat diet-and ovariectomy-induced obesity in mice [J]. J Biol Chem, 2010, 285: 31292-31303. |
| [37] | Giddabasappa A, Eswaraka JR, Barrett CM, et al. β-LGND2, an ERβ selective agonist, inhibits pathologic retinal neovascularization [J]. Invest Ophthalmol Vis Sci, 2012, 53: 5066-5075. |
| [38] | Pedram A, Razandi M, Narayanan R, et al. Estrogen regulates histone deacetylases to prevent cardiac hypertrophy [J]. Mol Biol Cell, 2013, 24: 3805-3818. |
| [39] | Möcklinghoff S, van Otterlo WAL, Rose R, et al. Design and evaluation of fragment-like estrogen receptor tetrahydroisoquinoline ligands from a scaffold-detection approach [J]. J Med Chem, 2011, 54: 2005-2011. |
| [40] | Manas ES, Unwalla RJ, Xu ZB, et al. Structure-based design of estrogen receptor-β selective ligands [J]. J Am Chem Soc, 2004, 126: 15106-15119. |
| [41] | Guo SW, Evers JLH. Lack of transparency of clinical trials on endometriosis [J]. Obstet Gynecol, 2013, 121: 1281-1290. |
| [42] | Sundén H, Ma JN, Hansen LK, et al. Design of a highly selective and potent class of non-planar estrogen receptor β agonists [J]. ChemMedChem, 2013, 8: 1283-1294. |
| [43] | De Angelis M, Stossi F, Carlson KE, et al. Indazole estrogens: highly selective ligands for the estrogen receptor β [J]. J Med Chem, 2005, 48: 1132-1144. |
| [44] | Chesworth R, Wessel MD, Heyden L, et al. Estrogen receptor beta selective ligands: discovery and SAR of novel heterocyclic ligands [J]. Bioorg Med Chem Lett, 2005, 15: 5562-5566. |
| [45] | Shiau AK, Barstad D, Radek J, et al. Structural characterization of subtype-selective ligand reveals a novel mode of estrogen receptor antagonism [J]. Nat Struct Biol, 2002, 9: 359-364. |
| [46] | Henke BR, Consler TG, Go N, et al. A new series of estrogen receptor modulators that display selectivity for estrogen receptor β [J]. J Med Chem, 2002, 45: 5492-5505. |