 2015, Vol. 50
2015, Vol. 50



2. 长江大学电信学院, 湖北 荆州 434023
2. College of Electronics and Information, Yangtze University, Jingzhou 434023, China
热休克蛋白 (heat shock proteins,HSPs) 是机体在应激 (病毒感染、缺氧、紫外线照射等) 状态下合成的一组蛋白质分子,故又称为应激蛋白。HSPs最初于1962年由遗传学家 Ritossa发现,它是一类在生物进化过程中高度保守并广泛存在于原核及真核生物中的蛋白质。热休克蛋白既是细胞应激反应的生物标志物,也是细胞内重要的分子伴侣蛋白[1, 2]。HSPs在机体内主要功能是参与维持其客户蛋白的正确折叠,使得蛋白能够形成生理功能所需构象,从而在调节蛋白合成与降解平衡及蛋白定位中发挥重要作用。根据同源程度及分子量大小,热休克蛋白主要分为5个家族[3]:HSP90家族 (83~90 kD)、HSP70家族 (66~78 kD)、HSP60家族,小分子HSP家族 (15~30 kD),此外还有分子质量在100~110 kD的大分子HSPs。它们在细胞中具有不同的定位,因而行使不同的细胞功能。其中HSP90家族是当前研究的热点,获得了广泛的关注。
HSP90在细胞内含量丰富,占细胞蛋白总量的1%~2%。HSP90在胞浆内主要以同源二聚体的形式存在,其单体包括3个主要的结构域: N末端结构域、中间结构域和C末端结构域。这3个结构域之间相互协同,发挥HSP90的分子伴侣功能。HSP90发挥作用依赖于ATP结合于N端的ATP酶结构域,ATP的结合和水解产生构象转换作用,调节其参与的多亚基复合物的装配。越来越多的研究表明,HSP90抑制剂有可能成为一类杀伤多种肿瘤细胞的有效药物[4, 5, 6]。
1.1 HSP90与肿瘤的关系作为哺乳动物细胞内含量最多的蛋白,HSP90对于其客户蛋白的装配、运转、折叠以及降解发挥着十分重要的作用。研究表明,抑制HSP90的功能可导致其客户蛋白通过泛素-蛋白酶复合体途径进行降解。已经报道的HSP90的客户蛋白有280多种,其中48种与细胞生长或信号转导相关。许多客户蛋白是致癌基因的表达产物,其中多个已是临床上明确的抗肿瘤药物作用靶点。部分客户蛋白与HSP90的亲和力排序如下: HER2 > 突变型EGFR > Raf-1 > Akt > 突变型BRAF > 野生型EGFR[7]。
HSP90调控的客户蛋白很多都是肿瘤发病通路中的原癌基因表达产物或重要的信号转导因子,与肿瘤的发生和发展关系密切,因此抑制HSP90可以从多环节多途径影响癌细胞生长和存活,避免了单靶点治疗可能产生的耐药性。研究表明,HSP90在肿瘤细胞中主要处于活化态,在正常细胞中则主要处于静默态。HSP90的活性受到共伴侣蛋白分子的调控,而在肿瘤细胞中的共伴侣蛋白的表达量很高,所以导致肿瘤细胞中的HSP90活性高于正常细胞。因此HSP90成为一个很有研发前景的抗肿瘤药物靶点[7]。
1.2 HSP90抑制剂的亚型选择性及其生物学效应人源HSP90有4种亚型,胞浆内有HSP90α和HSP90β两种,GRP94存在于内质网,TRAP1则存在于线粒 体基质内。现有研究认为这4个成员的作用方式几 乎是一致的,但因为它们本身在细胞内的定位不同,所以它们所结合的客户蛋白也不尽相同。胞浆内的HSP90α/β在肿瘤细胞中高表达,并且其客户蛋白大多都在肿瘤发生发展的信号转导通路、细胞周期调控和细胞凋亡路径中发挥着重要作用,比如HER2、EGFR、Raf-1、AKT等等。越来越多的证据表明,细胞器内的伴侣蛋白GRP94和TRAP1同样和肿瘤关系密切。GRP94的一个重要功能是维持分泌蛋白和膜蛋白的活性构象,其客户蛋白包括免疫球蛋白、TLRs和整合素类。肿瘤细胞中GRP94的高表达往往会导致细胞增殖和新陈代谢的速度加快以及耐药性的产生。TRAP1在肿瘤细胞中的高表达同样会导致多药耐药性的产生。在肿瘤细胞中,TRAP1和HSP90α/β一同发挥作用,保护肿瘤细胞不受氧化应激和细胞凋亡的影响,从而维持线粒体的完整性。近年来,越来越多的人开始关注HSP90抑制剂对不同亚型的选择性,因为不同的选择性往往会产生不同的生物学效应。Taldone等[8]综合运用计算机辅助药物设计和现有的复合物晶体结构,发现了除蛋白铰链区结合口袋之外的另外两个口袋,这些口袋不同的理化性质将决定抑制剂的亚型选择性。Duerfeldt等[9]采用基于结构的药物设计方法发现了一类对GRP94亲和力优于其他亚型的抑制剂。该类化合物在HEK293细胞中能够显著抑制TLRs向细胞膜的转运,但在MCF-7和SkBr3细胞中对于HSP90α/β的客户蛋白没有降解作用。Ernst等[10]发现的一类二氢吲哚酮类的化合物对HSP90α/β(Ki = 5 nmol·L-1) 的亲和力明显优于GRP94和TRAP1 (Ki > 10μmol·L-1)。该类化合物没 有明显的细胞毒作用,但对于Huntington舞蹈病有显著的治疗效果。近年研究表明,对于HSP90的调控对治疗神经退行性疾病也有显著疗效[10, 11]。
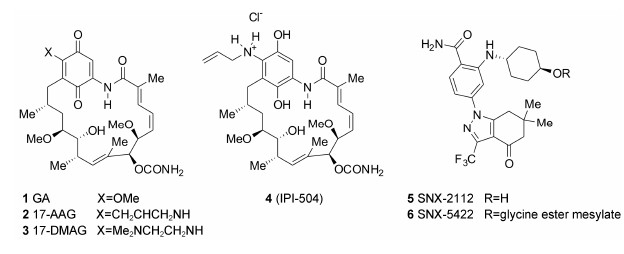
2 HSP90抑制剂简介自格尔德霉素 (geldanamycin,GA) 在1992年作为第一个HSP90抑制剂被发现以来[12],越来越多的其他结构类型的抑制剂被开发并陆续进入到临床研究阶段,但还没有作用于HSP90的靶向抑制剂作为抗肿瘤药物上市。目前,在研的HSP90抑制剂有1个处于临床III期 (STA-9090)、6个处于临床II期 (Debio-0932、KW-2478、AT-13387、SNX-5422、AUY- 922、17-AAG)、3个处于临床I期 (TAS-116、XL-888、PU-H71),还有49个化合物处于发现阶段 (图 1)。

|
Figure 1 The clinical statusof HSP90 inhibitors |
HSP90抑制剂根据其与蛋白结合位点的不同主要分为N端抑制剂、C端抑制剂和中间域抑制剂。作用于HSP90 C-端的抑制剂目前研究较少,现已发现的主要有没食子酸酯、新生霉素两类[13]。海洋天然产物sansalvamide A的衍生物sansalvamide A-amide (SanA-amide) 是目前报道的唯一一类结合于HSP90中间域结合位点的抑制剂,通过变构调节作用,抑制HSP90 C端和客户蛋白及共伴侣蛋白的结合,但对于N端和客户蛋白结合没有明显的影响[14]。目前进入临床研究的HSP90抑制剂均作用于HSP90 N端ATP结合位点。本文在已有的HSP90综述基础上[13],根据抑制剂与ATP结合口袋铰链区3种不同的结合模式 (图 2),重点介绍与临床研究密切相关的小分子化合物,综述N端的HSP90抑制剂的研发及目前的临床研究进展。
2.1 第一类小分子抑制剂以酰胺为关键结合基团与HSP90的氨基酸残基Asp93相结合格尔德霉素(geldanamycin,GA) 是最早被发现的HSP90抑制剂。它是在20世纪70年代从放射菌类肉汤中分离得到的天然产物,起初作为抗生素使用,直到20世纪80年代才发现该化合物具有抗肿瘤活性。格尔德霉素虽有明显的抗肿瘤效应,但也有较强的肝毒性,且在动物体内容易代谢失活,这些缺陷限制了其作为抗肿瘤药物的进一步研发[15, 16, 17, 18]。
人们成功研制出格尔德霉素类似物17-烯丙胺- 17-去甲氧基格尔德霉素 (17-AAG)。17-AAG是17位甲氧基被烯丙胺基取代后得到的GA衍生物,其抗肿瘤活性高于GA,而且肝毒性也大大降低,是第一个进入临床试验的HSP90抑制剂,目前已经进入II期临床试验阶段。但是17-AAG的水溶性较差,仍有一定的肝毒性且生物利用度有限,对不同类型的肿瘤和不同情况的患者疗效差异较大[19, 20]。
17-AAG的溶解性较差,通过制剂改善其生物利用度,效果不是很理想。研究表明,17-AAG的苯醌 可以互变成为对苯二酚,且后者对于HSP90有更强的抑制活性。IPI-504是17-AAG还原为苯酚的盐酸盐。分子模拟研究显示IPI-504与HSP90蛋白结合时不仅保持了构象,而且氢键的数量有所增加,所以比17-AAG具有更好的亲和力。IPI-504以盐酸盐的形式存在,具有更好的水溶性,并且降低了毒副作用 (肝毒性、心脏毒性、恶心、呕吐)[21]。
17-DMAG也是GA 17位取代的衍生物,其17位的2-甲基乙二胺基团伸向溶剂占据的结合腔的开口处,其水溶性和生物利用度有所增加,于2006年进入临床试验,但于2008年终止实验[22]。
SNX-2112是一类结构新颖的苯甲酰胺类小分子HSP90抑制剂,它是通过高通量筛选发现并优化获得的,具有良好的体内和体外活性 (HSP90 Ki = 16nmol·L-1,HER2 IC50 = 10nmol·L-1)[23]。其前药SNX- 5422(HSP90 Ki = 41 nmol·L-1,HER2 IC50 = 37 nmol·L-1) 于2007年5月底开展临床I期试验,评价其安全性。化合物SNX-5422的体内实验发现,无论采取口服还是静脉推注的给药方式,在3 h左右达峰,24 h基本代谢完全,在体内不产生蓄积作用。因此,SNX-5422采用隔天一次口服用药,具有良好的耐受性。SNX-5422主要用于对常规化疗无效的白血病、淋巴瘤、淋巴系统增生性疾病、小肠癌和非特异性的成人实体瘤的治疗,目前已经进入临床II期阶段。

|
Figure 2 The binding modes ofinhibitors in hingeregion of ATP pocket |
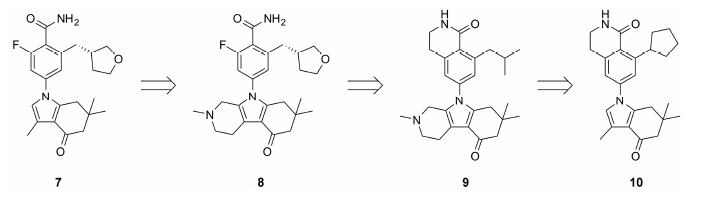
化合物7是2010年报道的一类对HSP90α/β有较好选择性的抑制剂,具有较好的血脑屏障透过率。为了发现具有更好的亚型选择性、细胞活性和高血 脑屏障透过率的先导化合物,Ernst等[10]首先将四氢吲哚酮部分替换为三环的四氢吲哚酮,化合物8的哌啶环很好地占据了螺旋104-111形成的疏水口袋,氮原子通过水分子和ASN51形成了新的氢键作用,哌啶环的碱性 (pKa = 7.6)使得化合物水溶性有了很大提高,解决了HSP90许多抑制剂 (包括四氢吲哚酮类) 溶解性差的问题。但是该化合物与7相比,亚型选择性并没有得到提高。通过分析化合物8苯甲酰 胺部分与HSP90四种亚型结合口袋的差异,作者发现在HSP90α/β中186号残基是缬氨酸,而在GRP94和TRAP1的相同位置是异亮氨酸,因此可以利用两者空间位阻的差异产生相应的选择性。作者认为在苯甲酰胺邻位上引入两个碳原子可以很好地与V186残基形成疏水作用,而与异亮氨酸形成位阻。为了避免邻位引入的取代基对酰胺所形成的关键氢键作用产生影响,作者将酰胺进行环化,这样不仅去除了1个不必要的氢键,而且降低了整个分子的极性表面 (PSA),增强了血脑屏障通透性。设计得到的化合物9具有很好的亚型选择性,对于细胞内亨廷顿蛋白突变体 (mutant Huntington protein) 有很好的清除率,对于HSP90的客户蛋白CDK4和Akt有明显的下调作用。对化合物9的PK性质研究发现其有较高的血浆清除率和脑内暴露量,并通过一些研究发现哌啶环是产生上述性质的重要结构因素,因此研究人员再次将三环的四氢吲哚酮替换为3-甲基四氢吲哚酮。化合物10对HSP90α/β的结合力显著强于GRP94和TRAP1,因此该类化合物不会对GRP94和TRAP1的客户蛋白产生影响,因而降低了相应的不良反应 (表 1)。化合物10对于突变性的mHtt有很强的降解、清除作用,细胞毒作用小,血脑屏障透过率高,在大鼠动物实验中显示了对于退行性神经疾病很好的治疗效果,目前该化合物已进入临床前研究。
|
|
Table 1 Isoform selectivity profilesof compounds 7-10 |
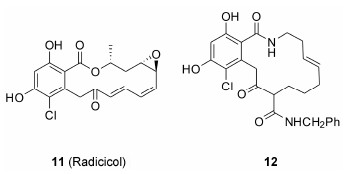
根赤壳菌素 (radicicol,RD)是从真菌Monosporiumbonorden分离得到的大环类抗生素,其作用位点也是HSP90的N-端的ATP结合区域,在体外对HSP90的亲和力比 GA和17-AAG高 (Kd = 19nmol·L-1)。但在体内,由于根赤壳菌素含有亲电子环氧环和Michael受体,易与含巯基的亲核试剂发生麦氏加成反应失去抗肿瘤活性[24]。Radicicol虽然成药性较差,但其具有的双羟基结构类型为之后大量新型抑制剂的设计提供了模板和思路。
Day等[25]发现了一系列大环内酰胺类的radicicol类似物。化合物12在人肝微粒体中的代谢清除率为47%,相较于radicicol (83%) 的稳定性有了明显提 高,而且该化合物比其他类型的radicicol类似物对 人结肠癌细胞有更强的抑制活性 (HCT116: 12 IC50 = 600nmol·L-1,RD IC50 = 7 600nmol·L-1)。
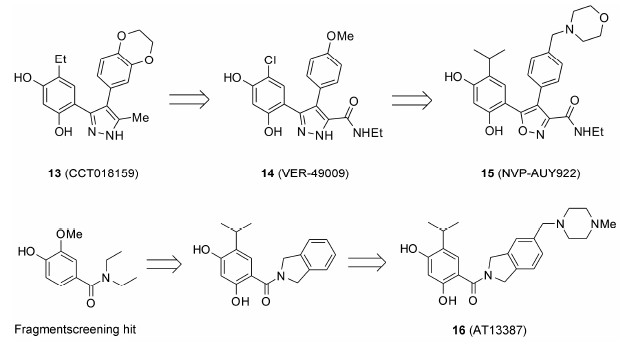
Workman等[26, 27, 28, 29]通过高通量筛选得到的化合物CCT018159,在酵母菌HSP90的ATP酶活性实验中,其IC50为7.1 μmol·L-1。在此基础上,利用基于结构药物分子设计策略,对其进行优化得到VER-49009,在体外的抑制活性和17-AAG相当。对VER-49009继续进行结构优化产生了异噁唑-间苯二酚类似物NVP-AUY922(后来被Novartis公司收购)[30, 31],保持了与HSP90结合的氢键网络,避免了吡唑环的互变异构,异丙基的引入增强了细胞活性。NVP-AUY922已经进入临床II期试验阶段,主要针对HER2阳性或ER阳性的晚期或转移性乳腺癌患者。17-DMAG仅对醌氧化还原酶高水平表达的肿瘤细胞有效,而醌氧化还原酶基因缺失或突变的肿瘤细胞对17-DMAG具有抵抗。异噁唑类骨架的HSP90抑制剂则不依赖醌氧化还原酶,从而具有更广泛的应用前景。通过借鉴AUY922的结合模式,大量的含有间苯二酚结构的抑制剂被报道。
Astex公司通过基于片段的药物设计方法,发现了一类具有二氢异吲哚结构的化合物AT13387[32, 33, 34, 35]。该化合物在低浓度下对于多种胃肠道基质肿瘤细胞有很好的增殖抑制活性。通过引入甲基哌嗪,化合物不仅活性能够保持,生物利用度也有了较大提高。目前该化合物正在进行临床I/II期试验,主要针对前列腺癌、非小细胞肺癌,以及和达拉菲尼、曲美替尼联合用药治疗晚期黑色素瘤。
STA9090是一类含有三唑环的间苯二酚类化合物,对于HSP90的调控、抑制有着非常明显的作用。该化合物目前正在针对多种恶性肿瘤进行临床I/II期试验,包括去势抵抗性前列腺癌、小细胞肺癌、黑色素瘤等。在用于晚期非小细胞肺癌的II期试验中获得了肯定性结果,显示出了该化合物具有缩小肿瘤与延缓疾病进程的作用。目前STA9090与多烯紫杉醇联合用药治疗肺腺癌的试验已经进入到临床III期阶段[5]。
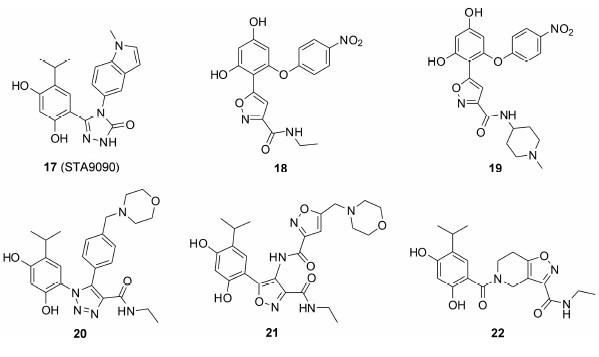
化合物18是Brasca等[36]通过基于片段的药物设计方法发现的一类结构,对HSP90的结合活性IC50为48 nmol·L-1,对人肿瘤细胞A2780的增殖抑制活性IC50为80 nmol·L-1,酰胺上二级胺氮原子和残基Gly97、Lys58形成很强的氢键作用,对于活性保持 十分关键。在酰胺部分引入位阻更大的水溶性基团 氮甲基哌嗪后,化合物19在分子和细胞水平活性都有所提高 (HSP90 IC50 = 10nmol·L-1,A2780 IC50 = 69nmol·L-1)。化合物20是一类含有三氮唑结构的分子,其分子和细胞水平都有较强的活性[37] (HSP90 IC50 < 5 nmol·L-1,NCI-H460 IC50 = 4nmol·L-1)。Baruchello等[38, 39]报道了两类含有异噁唑环的分子,化合物21虽然在体外对于非小细胞肺癌的IC50只有200 nmol·L-1,但在体内对于人类鳞状细胞癌移植瘤模型,腹腔给药60mg·kg-1有很好的抑瘤效果 (抑瘤率48%)。化合物22与HSP90的结合力较强 (IC50 = 29nmol·L-1),但对NCI-H460细胞的增殖抑制能力较弱 (IC50 = 450nmol·L-1)。
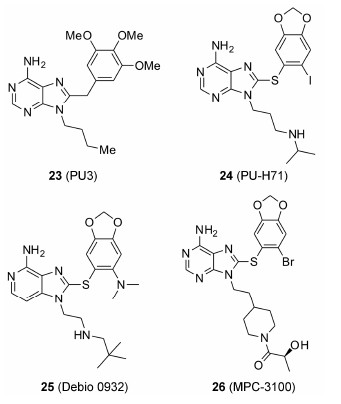
2001年,Wright等[40]报道了一种以嘌呤为母核的HSP90抑制剂PU3,这是第一个化学合成的HSP90小分子抑制剂。该分子是通过计算机辅助药物设计,将ATP的腺嘌呤环和17-AAG的芳香环部分进行组合拼接,进而得到了一类嵌合的分子。PU3的作用位点与GA一致,均为HSP90N-端ATP结合位点,几乎能占据HSP90的ATP结合口袋中所有重要的作用位点。它在抑制HSP90受体蛋白降解和抗肿瘤能力方面类似GA,但是活性较弱 (HER2 IC50 = 40 mmol·L-1)。Conforma公司[41, 42, 43]和Sloan-Kettering研究所[44]分别对这类 结构进行了优化,并得到了一系列活性在30~100nmol·L-1之间的化合物。其中化合物24 (PU-H71) 对HSP90结合活性为51 nmol·L-1,对三阴性乳腺癌(TNBC) 细胞系MDA-MB-468、MDA-MB-231和HCC-1806,IC50分别为65、140和87 nmol·L-1,目前该化合物正在进行I期临床试验。该类化合物以PU-H71为例,构效关系分析如下: ① 6位氨基和1位氮原子和Asp93残基形成很强的氢键网络作用,7位氮原子与保守水分子形成氢键作用; ② 富电子的苯环处于π-π作用区,和Phe138形成π-π堆积作用;③ 9位氮原子的侧链伸向溶剂区,这一部分对于结合活性和分子的理化性质都有影响; ④ hinge区和π-π作用区必须通过适当的基团进行连接,保持两部分处于C型构象来发挥作用,当连接基团为S原子、亚甲基时活性相当,但如果换成O、N原子则活性丧失 (图 3)。

|
Figure 3 The binding mode of PU-H71 |
很多研究机构对PU这类结构进行了大量改造,代表性的化合物为25和26。化合物25 (Debio 0932) 于2010年进入临床I期试验,治疗晚期实体瘤和淋巴瘤,取得了很好的效果,并于2012年进入临床II期试验[45]。化合物26 (MPC-3100) 是通过对PU系列化合物N-9侧链的改造,发现的一类N-甲酰基-4-哌啶的结构。构效关系显示N-9和哌啶环之间为两个碳原子时活性最好,去除甲酰基或改用烷基取代活性变弱。化合物26具有较好的分子水平和细胞水平活性 (HSP90 IC50 = 140nmol·L-1; HCT116 IC50 = 540nmol·L-1)、合理的血浆稳定性和PK性质[46]。
基于PU-H71的作用模式,Kasibhatla等[47]通过骨架跃迁,将NH2和Ar进行“扭转”,将两者的距离保持在5Å,得到了一类新颖的结构。因为Ar的位置由C-8移到了N-9,合成的步骤大大简化。经过后续的一系列分子优化和活性评价,发现了化合物27 (HER2 IC50 = 30 nmol·L-1,MCF7 IC50 = 100nmol·L-1,BT474 IC50 = 100nmol·L-1) (图 4)。

|
Figure 4 The discovery process of BIIB021 |
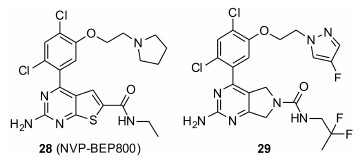
NVP-BEP800是Vernalis公司2009年报道的一类结构新颖的分子[48]。研究人员分别通过计算机辅助药物设计和基于片段的药物发现方法发现了有微弱活性的分子片段,通过将这些片段进行组合拼接以及后续的结构优化,发现一系列活性在50~100nmol·L-1之间的化合物。代表性化合物28 (NVP- BEP800) 对HSP90活性为58 nmol·L-1,对HCT116、MCF7、PC3M、A375、T24、U87MG肿瘤细胞的GI50分别为0.12、0.11、1.05、0.038、0.145、0.875 μmol·L-1。Pfizer公司于2011年发现的化合物29和28具有相似的结构,但活性更强[49](HSP90 Ki = 7 nmol·L-1,A2058 IC50 = 20nmol·L-1,A375 IC50 = 40nmol·L-1,HT-29 IC50 = 3nmol·L-1)。
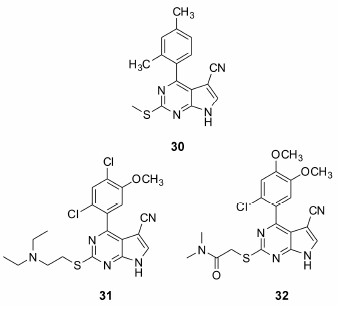
Davies等[50]通过基于片段的药物发现方法发现了一类氰基吡咯[2,3-d]嘧啶的结构。化合物30具有中等的结合活性 (HSP90 IC50= 200 nmol·L-1)。氰基取代了一个保守的水分子,并且和Asn51残基形成了氢键作用。后续的优化发现了化合物31 (HSP90 IC50 < 1 nmol·L-1,BT474 GI50 = 72 nmol·L-1) 和32 (HSP90 IC50 < 1 nmol·L-1,BT474 GI50 = 34 nmol·L-1),但是32的心脏毒性更小 (hERG IC50 > 30μmol·L-1)。
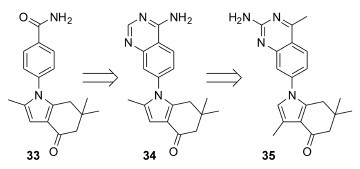
Serenex公司的研究人员将其之前发现的酰胺类化合物33通过骨架跃迁发现了化合物34,有一定的细胞活性,结合之前的构效关系研究,以及分子模拟,得到了高活性的化合物35[51] (HSP90 IC50 = 98nmol·L-1,AU565 IC50 = 4nmol·L-1,MCF IC50 = 7nmol·L-1; HT29 IC50 = 5nmol·L-1)。
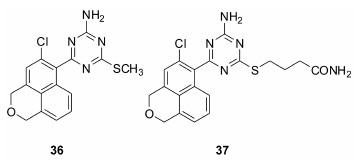
Suda等[52, 53]发现了以2-氨基-1,3,5-三嗪结构为母核的化合物36,对HSP90的结合活性达到了3.4 nmol·L-1,对不同的肿瘤细胞也有较好的抑制活性(HCT116,IC50= 460 nmol·L-1; NCI-N87 IC50 = 570nmol·L-1)。然而该化合物的溶解性较差,口服生物利用度较低。将甲硫基替换为丁基酰胺得到的化合物37,不仅对HSP90的结合能力更强 (IC50 = 0.48 nmol·L-1), 水溶性和生物利用度也有了很大提高。
HSP90的生物学、药理学、药物化学研究依然是抗肿瘤新药研发和临床研究的热点领域。目前,通过生物标记物方法,探索进入临床研究化合物的药效学正在进行中,但这些化合物均未考虑HSP90亚型选择性,因而可能会产生心脏毒性和胃肠道等不良反应。随着对HSP90所调控的细胞通路更深入的认识,才能更得心应手地通过对HSP90的不同亚型的选择性调控,达到更好的治疗疾病的效果。药物化学家目前正致力于解决HSP90抑制剂的亚型选择性,但由于哺乳动物类HSP90的结构高度同源性,也许需要通过寻找ATP结合口袋以外的其他关键结合位点来解决这一问题。随着对HSP90分子作用机制研究的深入以及临床评价手段的日趋完善,HSP90抑制剂必将在包括肿瘤在内的多个治疗领域展示出良好的应用前景。
| [1] | Welch WJ. Mammslian stress response: cell physiology structure/function of stress proteins, and implications for medicine and disease [J]. Physiol Rev, 1992, 72: 1063-1081. |
| [2] | Burdon RH. Heat shock proteins in relation to medicine [J]. Mol Asp Med, 1993, 14: 83-165. |
| [3] | Bao XQ, Liu GT. Heat shock proteins: new target in cytoprotective and tumor therapy [J]. Acta Pharm Sin (药学学报), 2008, 43: 234-240. |
| [4] | Rutherford SL, Lindquist S. HSP90 as a capacitor for morphological evolution [J]. Nature, 1998, 396: 336-342. |
| [5] | Bhat R, Tummalapalli SR, Rotella DP. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors [J]. J Med Chem, 2014, 57: 8718-8728. |
| [6] | Chen Y, Ding J. Heat shock protein 90: novel target for cancer therapy [J]. Chin J Cancer, 2004, 23: 968-974. |
| [7] | Biamonte MA, Van-de-Water R, Arndt JW, et al. Heat shock protein 90: inhibitors in clinical trials [J]. J Med Chem, 2010, 53: 3-17. |
| [8] | Taldone T, Patel PD, Patel M, et al. Experimental and structural testing module to analyze paralog-specificity and affinity in the Hsp90 inhibitors series [J]. J Med Chem, 2013, 56: 6803-6818. |
| [9] | Duerfeldt AS, Peterson LB, Maynard JC, et al. Development of a Grp94 inhibitor [J]. J Am Chem Soc, 2012, 134: 9796-9804. |
| [10] | Ernst JT, Neubert T, Liu M, et al. Identification of novel Hsp90α/β isoform selective inhibitors using structure-based drug design. Demonstration of potential utility in treating CNS disorders such as Huntington's disease [J]. J Med Chem, 2014, 57: 3382-3400. |
| [11] | Paul S, Mahanta S. Association of heat-shock proteins in various neurodegenerative disorders: is it a master key to open the therapeutic door? [J]. Mol Cell Biochem, 2014, 386: 45-61. |
| [12] | Whitesell L, Shifrin SD, Schwab G, et al. Benzoquinonoid ansamycins possess selective tumoricidal activity unrelated to src kinase inhibition [J]. Cancer Res, 1992, 52: 1721-1728. |
| [13] | Luo HM, Sun W, Yin JY, et al. Progress in the study of heat shock protein 90 inhibitors [J]. Acta Pharm Sin (药学学报), 2010, 45: 813-820. |
| [14] | Vasko RC, Rodriguez RA, Cunningham CN, et al. Mechanistic studies of sansalvamide A-amide: an allosteric modulator of Hsp90 [J]. ACS Med Chem Lett, 2010, 1: 4-8. |
| [15] | Liao ZY, Zhen YS. Advances in antitumor activity of the Hsp90 inhibitor geldanamycin [J]. Acta Pharm Sin (药学学报), 2001, 36: 716-720. |
| [16] | Roe SM, Prodromou C, O'Brien R, et al. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin [J]. J Med Chem, 1999, 42: 260-266. |
| [17] | Jez JM, Chen JCH, Rastelli G, et al. Crystal structure and molecular modeling of 17-DMAG in complex with human Hsp90 [J]. Chem Biol, 2003, 10: 361-368. |
| [18] | Shen G, Wang M, Welch TR, et al. Design, synthesis, and structure activity relationships for chimeric inhibitors of Hsp90 [J]. J Org Chem, 2006, 71: 7618-7631. |
| [19] | Agnew EB, Wilson RH, Grem JL, et al. Measurement of the novel antitumor agent 17-(allylamino)-17-demethoxygeldanamycin in human plasma by high-performance liquid chromatography [J]. J Chromatogr B Biomed Sci Appl, 2001, 755: 237-243. |
| [20] | Solit DB, Zheng FF, Drobnjak M, et al. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts [J]. Clin Cancer Res, 2002, 8: 986-993. |
| [21] | Hanson BE, Vesole DH. Retaspimycin hydrochloride (IPI-504): a novel heat shock protein inhibitor as an anticancer agent [J]. Expert Opin Invest Drugs, 2009, 18: 1375-1383. |
| [22] | Jhaveri K, Taldone T, Modi S, et al. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers [J]. Biochim Biophys Acta (BBA)-Mol Cell Res, 2012, 1823: 742-755. |
| [23] | Huang KH, Veal JM, Fadden RP, et al. Discovery of novel 2-aminobenzamide inhibitors of heat shock protein 90 as potent, selective and orally active antitumor agents [J]. J Med Chem, 2009, 52: 4288-4305. |
| [24] | Kwon HJ, Yoshida M, Abe K, et al. Radicicol, an agent inducing the reversal of transformed phenotypes of src-transformed fibroblasts [J]. Biosci Biotechnol Biochem, 1992, 56: 538-539. |
| [25] | Day JEH, Sharp SY, Rowlands MG, et al. Targeting the Hsp90 molecular chaperone with novel macrolactams. Synthesis, structural, binding, and cellular studies [J]. ACS Chem Biol, 2011, 6: 1339-1347. |
| [26] | Barril X, Beswick MC, Collier A, et al. 4-Amino derivatives of the Hsp90 inhibitor CCT018159 [J]. Bioorg Med Chem Lett, 2006, 16: 2543-2548. |
| [27] | Brough PA, Barril X, Beswick M, et al. 3-(5-Chloro-2, 4-dihydroxyphenyl)-pyrazole-4-carboxamides as inhibitors of the Hsp90 molecular chaperone [J]. Bioorg Med Chem Lett, 2005, 15: 5197-5201. |
| [28] | Cheung KMJ, Matthews TP, James K, et al. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3, 4-diarylpyrazole class of Hsp90 inhibitors [J]. Bioorg Med Chem Lett, 2005, 15: 3338-3343. |
| [29] | Dymock BW, Barril X, Brough PA, et al. Novel, potent small-molecule inhibitors of the molecular chaperone Hsp90 discovered through structure-based design [J]. J Med Chem, 2005, 48: 4212-4215. |
| [30] | Brough PA, Aherne W, Barril X, et al. 4, 5-Diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer [J]. J Med Chem, 2008, 51: 196-218. |
| [31] | Sharp SY, Boxall K, Rowlands M, et al. In vitro biological characterization of a novel, synthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors [J]. Cancer Res, 2007, 67: 2206-2216. |
| [32] | Kung PP, Huang B, Zhang G, et al. Dihydroxyphenylisoindoline amides as orally bioavailable inhibitors of the heat shock protein 90 (Hsp90) molecular chaperone [J]. J Med Chem, 2009, 53: 499-503. |
| [33] | Kung PP, Funk L, Meng J, et al. Dihydroxylphenyl amides as inhibitors of the Hsp90 molecular chaperone [J]. Bioorg Med Chem Lett, 2008, 18: 6273-6278. |
| [34] | Murray CW, Carr MG, Callaghan O, et al. Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency [J]. J Med Chem, 2010, 53: 5942-5955. |
| [35] | Woodhead AJ, Angove H, Carr MG, et al. Discovery of (2, 4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1, 3-dihydroisoindol-2-yl] methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design [J]. J Med Chem, 2010, 53: 5956-5969. |
| [36] | Brasca MG, Mantegani S, Amboldi N, et al. Discovery of NMS-E973 as novel, selective and potent inhibitor of heat shock protein 90 (Hsp90) [J]. Bioorg Med Chem, 2013, 21: 7047-7063. |
| [37] | Taddei M, Ferrini S, Giannotti L, et al. Synthesis and evaluation of new Hsp90 inhibitors based on a 1, 4, 5-trisubstituted 1, 2, 3-triazole scaffold [J]. J Med Chem, 2014, 57: 2258-2274. |
| [38] | Baruchello R, Simoni D, Grisolia G, et al. Novel 3, 4-isoxazolediamides as potent inhibitors of chaperone heat shock protein 90 [J]. J Med Chem, 2011, 54: 8592-8604. |
| [39] | Baruchello R, Simoni D, Marchetti P, et al. 4, 5, 6, 7-Tetrahydro-isoxazolo-[4, 5-c]-pyridines as a new class of cytotoxic Hsp90 inhibitors [J]. Eur J Med Chem, 2014, 76: 53-60. |
| [40] | Wright L, Barril X, Dymock B, et al. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms [J]. Chem Biol, 2004, 11: 775-785. |
| [41] | Kasibhatla SR, Hong K, Zhang L, et al. Preparation of purine analogs as heat shock protein 90 (HSP90) inhibitors: WO 03/037860 [P]. 2003. |
| [42] | Biamonte MA, Shi J, Hong K, et al. Orally active purine-based inhibitors of the heat shock protein 90 [J]. J Med Chem, 2006, 49: 817-828. |
| [43] | Zhang L, Fan J, Vu K, et al. 7'-Substituted benzothiazolothio-and pyridinothiazolothio-purines as potent heat shock protein 90 inhibitors [J]. J Med Chem, 2006, 49: 5352-5362. |
| [44] | Llauger L, He H, Kim J, et al. Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90 [J]. J Med Chem, 2005, 48: 2892-2905. |
| [45] | Bao R, Lai CJ, Qu H, et al. CUDC-305, a novel synthetic HSP90 inhibitor with unique pharmacologic properties for cancer therapy [J]. Clin Cancer Res, 2009, 15: 4046-4057. |
| [46] | Kim SH, Bajji A, Tangallapally R, et al. Discovery of (2S)-1-[4-(2-{6-amino-8-[(6-bromo-1, 3-benzodioxol-5-yl) sulfanyl]-9H-purin-9-yl} ethyl) piperidin-1-yl]-2-hydroxypropan-1-one (MPC-3100), a purine-based Hsp90 inhibitor [J]. J Med Chem, 2012, 55: 7480-7501. |
| [47] | Kasibhatla SR, Hong K, Biamonte MA, et al. Rationally designed high-affinity 2-amino-6-halopurine heat shock protein 90 inhibitors that exhibit potent antitumor activity [J]. J Med Chem, 2007, 50: 2767-2778. |
| [48] | Brough PA, Barril X, Borgognoni J, et al. Combining hit identification strategies: fragment-based and in silico approaches to orally active 2-aminothieno [2, 3-d] pyrimidine inhibitors of the Hsp90 molecular chaperone [J]. J Med Chem, 2009, 52: 4794-4809. |
| [49] | Zehnder L, Bennett M, Meng J, et al. Optimization of potent, selective, and orally bioavailable pyrrolodinopyrimidine-containing inhibitors of heat shock protein 90. Identification of development candidate 2-amino-4-{4-chloro-2-[2-(4-fluoro-1H-pyrazol-1-yl)ethoxy]-6-methylphenyl}-N-(2, 2-difluoropropyl)-5, 7-dihydro-6H-pyrrolo [3, 4-d] pyrimidine-6-carboxamide [J]. J Med Chem, 2011, 54: 3368-3385. |
| [50] | Davies NGM, Browne H, Davis B, et al. Targeting conserved water molecules: design of 4-aryl-5-cyanopyrrolo [2, 3-d] pyrimidine Hsp90 inhibitors using fragment-based screening and structure-based optimization [J]. Bioorg Med Chem, 2012, 20: 6770-6789. |
| [51] | Huang KH, Barta TE, Rice JW, et al. Discovery of novel aminoquinazolin-7-yl 6, 7-dihydro-indol-4-ones as potent, selective inhibitors of heat shock protein 90 [J]. Bioorg Med Chem Lett, 2012, 22: 2550-2554. |
| [52] | Miura T, Fukami TA, Hasegawa K, et al. Lead generation of heat shock protein 90 inhibitors by a combination of fragment-based approach, virtual screening, and structure-based drug design [J]. Bioorg Med Chem Lett, 2011, 21: 5778-5783. |
| [53] | Suda A, Kawasaki K, Komiyama S, et al. Design and synthesis of 2-amino-6-(1H, 3H-benzo[de]-isochromen-6-yl)-1, 3, 5-triazines as novel Hsp90 inhibitors [J]. Bioorg Med Chem, 2014, 22: 892-905. |