2015, Vol. 50
2015, Vol. 50

坎格列净 (canagliflozin) 是以2型钠葡萄糖共转运蛋白 (SGLT2) 为靶标的第一个口服降血糖药, 通过可逆的选择性抑制肾小管对血糖的重吸收, 促进血糖在尿液中的排泄, 降低体内血糖水平, 因而作用机制有别于已有降血糖药物。坎格列净也是以天然活性产物为先导物研制成功的范例。 1 靶标特点
生理学研究表明, 循环血中的葡萄糖在肾小球中滤过, 然后在肾脏近曲小管处重吸收, 重吸收作用是由两个蛋白介导: 1型和2型钠葡萄糖共转运蛋白 (SGLT1和SGLT2)。SGLT1是高选择性低容量的转 运蛋白, 主要在小肠上表达; SGLT2是低选择性高容量的转运蛋白, 主要表达于肾近曲小管的S1和S2区段上。SGLT2基因突变可导致持续的肾性糖尿。选择性的抑制SGLT2而不抑制SGLT1, 可成为不影响胃肠道吸收葡萄糖、不干预胰岛素系统的治疗2型糖尿病的新途径。 2 天然产物的初始改造
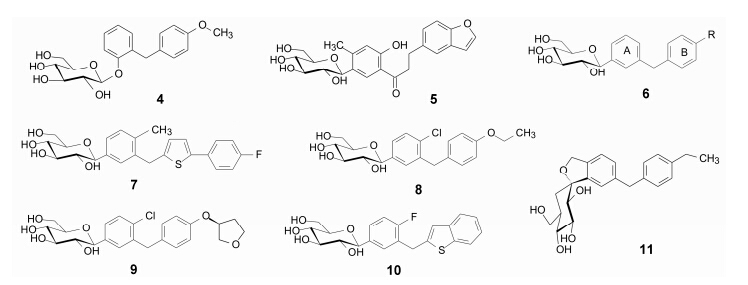
根皮苷 (1, phlorizin) 是以二氢查尔酮为苷元的葡萄糖苷, 含于许多果实中, 已知有150年的历史 (Rossetti L, et al. J Clin Invest, 1987, 79: 1510)。根皮苷具有抑制SGLT2和SGLT1的双重活性 (Toggenburger G, et al. Biochim Biophys Acta, 1982, 688: 557), 因选择性不高和在肠道迅速水解成根皮素 (phloretin) 和葡萄糖而失效, 根皮苷本身不能药用, 常作为药理工具药, 也是个良好的先导化合物。
根皮苷结构改造的目标是: ① 对SGLT2有高选择性抑制作用; ② 口服有效; ③ 消除糖苷容易水解失活的代谢不稳定性; ④ 化学结构具有新颖性。
初步的结构变换揭示出如下的构效关系: 糖基和两个苯环之间的连接基是必须的; A环的酚羟基可烷基化 (如甲氧基, 化合物2) 仍保持活性。A环换成苯并呋喃环, B环引入甲基得到的化合物3 (T-1095) 提高了选择性作用, 同等剂量下灌胃小鼠, 尿中葡萄 糖排泄量最大 (Tsujihara K, et al. Chem Pharm Bull, 1996, 44: 1174; Tsujihara K, et al. J Med Chem, 1999, 42: 5311; Oku A, et al. Diabetes, 1999, 48: 1794)。T-1095为前药 (碳酸酯), 仍未能克服O-糖苷的不稳定性。
与此同时, BMS公司也在以根皮苷为先导物研制SGLT2抑制剂, 将两个苯环间3个原子的连接基减少为1个, 成为类型4的化合物, 仍保持对SGLT2的选择性作用 (Washburn WN, et al. Chem Abstr, 2001, 135; 273163), 提示对先导物根皮苷的骨架可以作较大的变换。 3 C-糖苷提高稳定性
将3和4的O-苷换为C-苷, 使糖基经C-C键与苷元连接, 化合物5 (Tomiyama H, et al. Chem Abstr, 2001, 135: 304104) 和通式6 (Ellsworth B, et al. Chem Abstr, 2001, 134: 281069) 仍保持活性和选择性, 其代谢和化学稳定性显著强于相应的O-苷化合物3和4。 4 芳环的变换——杂环的引入和候选药物坎格列净的确定
为了优化活性和选择性以及实现结构的新颖性, 对6的A和B环分别用杂环做电子等排置换, 例如 A环用噻吩、吡咯、吡啶、吡嗪等杂环, B环用吡啶、吡嗪、吲哚、苯并呋喃、苯并噻吩、苯并噻唑、苯并噁唑、苯并咪唑等代替, 对化合物体外测定对人SGLT2 (hSGLT2) 的抑制活性 (IC50) 和对hSGLT1的选择性倍数。结果表明, A为苯环、B为苯基噻吩的母核活性和选择性优于其他系列, 其中化合物7 对hSGLT2和hSGLT1的IC50分别为2.2 nmol·L−1和910 nmol·L−1, 选择性高达414倍, 表明对小肠吸收葡萄糖的影响很小。大鼠口服生物利用度F = 83%, 血浆半衰期t1/2 = 5 h。雄性SD大鼠一次灌胃30 mg·kg−1, 尿中排泄3 696 mg葡萄糖/200 g体重。7被命名为 坎格列净 (canagliflozin) 进入临床前和临床研究 (Nomura S, et al. J Med Chem, 2010, 53: 6355)。强生 公司经III期临床研究表明, 坎格列净不仅显著降低 2型糖尿病患者的血糖水平, 而且极少引起低血糖事件。此外, 其减肥效果也十分明显。坎格列净于2013年经美国FDA批准上市 (Schernthaner G, et al. Diabetes Care, 2013, 36: 2508)。

达格列净 (8, dapagliflozin) 是BMS与AstraZeneca联合研制的第二个SGLT2抑制剂, 已于2014年经FDA批准上市 (Meng W, et al. J Med Chem, 2008, 51: 114)。作为C-糖苷可视作坎格列净的噻吩环被苯环替换的类似物, 为选择性作用更强的化合物, 对SGLT2和SGLT1的IC50分别为1.1 nmol·L−1和1 390 nmol·L−1。
由Boehlinger Ingrehaim与Eli Lilly联合研发的艾帕列净 (9, empagliflozin) 是第3个于2014年上市的SGLT2抑制剂, 也是C-葡萄糖苷, 艾帕列净与达格列净骨架结构相同, 只是将外侧苯环的乙基改为四氢呋喃片段, 对SGLT2的IC50为3.1 nmol·L−1, 作用于SGLT-1, 比4、5和6选择性高300倍以上 (Grempler R, et al. Diabetes Obes Metab, 2012, 14: 83−90)。
化合物10称作ipragliflozin, 是由Astellas和Merck联合研发的另一个SGLT2抑制剂, 也是C-葡萄糖苷, 结构中含有苯并噻吩环, 相当于坎格列净的噻吩与苯的并合。对SGLT2的抑制活性IC50 = 7.4 nmol·L−1, 强于SGLT1约254倍。大鼠灌胃的生物利用度F = 71.7%, 血浆半衰期t1/2 = 3.6 h。现处于III期临床研究 (Imamura M, et al. Bioorg Med Chem, 2012, 20: 3263)。
日本中外制药与韩国研发的tofogliflozin (11), 是将葡萄糖环换成多羟基环己烷与苯并二氢呋喃形成螺环化合物, 对hSGLT2和hSGLT1的IC50分别为 2.9 nmol·L−1和8 444 nmol·L−1, 选择性近3000倍, 对猴的生物利用度F = 85%。目前也在III期临床阶段(Ohtake Y, et al. J Med Chem, 2012, 55: 7828)。