2015, Vol. 50
2015, Vol. 50

西咪替丁是组胺H2受体第一个上市的拮抗剂, 通过抑制胃酸的分泌, 治疗胃和十二指肠溃疡。该药由Smith Kline & Frech研制, 于1976年在英国上市, 1979年FDA批准在美国上市。西咪替丁当时作为全球重磅药物是第一个通过理性药物设计 (rational drug design) 研制成功的药物, 主要研制者James Black爵士因本发明和之前发明的propranolol获得1988年诺贝尔生理和医学奖。
早在1964年就已知组胺可刺激胃壁细胞分泌胃酸, 然而抗组胺类的抗过敏药物不能阻断这个过程, 推测体内存在着不同于组胺引起过敏反应 (H1受体) 的另一种受体。
当时在对H2受体的结构和特征无所知晓的情况下, 研究拮抗H2受体的策略, 是寻找与组胺竞争性的结合受体的化合物, 以阻断组胺刺激胃酸分泌的功能。这样的化合物结构应类似组胺, 从而可被H2受体识别与结合, 但不刺激胃酸分泌。为此, 研究者从天然配体组胺开始, 组胺作为唯一的先导物, 经药物化学的设计-合成-活性评价-构效关系的理性设计, 成就了这一开创性的治疗药物。
首先, 项目以模拟组胺 (1) 的化学结构开始, 应用药物化学中的电子等排和同系原理, 在咪唑环、亚乙基链和伯胺进行结构变换, 合成了数百个组胺类似物, 寻找有拮抗活性的苗头化合物。苗头的发现并非一次完成, 而是通过分析和对比化合物结构、物化性质、激动与拮抗作用的表现逐步演化而成。
与组胺比较, 2-甲基组胺 (2) 刺激豚鼠回肠作用 (H1受体激动作用) 为组胺的17%, 但只有很弱的刺激胃酸分泌作用 (H2受体激动作用); 而4-甲基组胺 (3) 对回肠的刺激作用只为组胺的0.2%, 刺激胃酸分泌为组胺的50% (Durant et al. J Med Chem, 1976, 19: 923), 这说明咪唑环上引入甲基并变换甲基的位置可引起对H1和H2受体结合的显著变化。4-甲基组胺的结构与电性分布的低能优势构象, 扩大了激动H1和H2受体的差异, 大约250倍, 提示改变咪唑环和侧链的结构有可能提高对一种受体亚型的选择性作用。如 果化合物能够被H2受体选择性地识别和结合, 但不能使其活化, 应为H2受体拮抗剂。
评价化合物对胃酸分泌的作用是给实验动物以大剂量组胺, 使胃酸达到最大的分泌量, 然后给受试化合物测定胃酸量, 计算抑制活性。这种生理表型的评价方法虽然工作量巨大, 但更接近临床应用的状态。
另一方面, 变换侧链的氨基成胍基, 发现胍乙咪唑 (4) 有弱抑制胃酸分泌作用 (Durant GJ, et al. J Med Chem, 1975, 18: 830)。在pH 7.4生理状态下, 虽然氨基和胍基都可被质子化而带有正电菏, 但胍基的正电荷分布在4个杂化原子的轨道上, 分散在较大的平面范围内, 推测较广泛的正电荷结合区域有利于产生拮抗作用, 4的正电荷与咪唑环之间的距离大于组胺的氨基与咪唑的距离, 预示加长侧链有利于拮抗作用。4对H2受体仍有部分激动作用。
为了消除4对H2受体的激动作用, 一方面将胍基的非端基N换成S原子, 成为异硫脲化合物5, 目的是使正电荷只分散在两个外端N原子上, 结果表明对H2的拮抗作用进一步增强。另一方面加长4的侧链成胍丙咪唑 (6), 6抑制胃酸分泌作用比化合物4强6倍。然而5和6仍有激动剂作用。
将5和6的提高拮抗作用的因素合并, 以调整 末端基团的碱性和与咪唑环间的距离, 设计了侧链为3~5个碳原子的硫脲化合物, 在合成的众多化合物中, N-甲基取代的硫脲丁基咪唑 (7) 消除了激动 作用, 成为第一个H2受体拮抗剂, 称作布立马胺 (7, burimamide), 但布立马胺的药效活 性不高, 口服生物利用度低。布立马胺可认为是研发H2受体拮抗剂的先导化合物。

优化的路径怎样走, 研究者分析了布立马胺的结构, 咪唑环和硫脲基在生理pH条件下都可以发生互变异构: 咪唑环以3种共振形式存在 (图 1A), 硫脲基可采取4种不同的构象 (图 1B), 柔性碳链更有无数构象, 这可能是布立马胺药效构象少、活性不高的原因 (Black JW, et al. Nature, 1974, 248: 65; Emmett JC, et al. Inflam Res, 1979, 9: 26)。

|
图1 咪唑的互变异构 (A) 和甲基硫脲的不同构象 (B) |
咪唑的pKa(H) = 6.80, 组胺的咪唑环的pKa(H) = 5.90, 说明组胺的侧链为拉电子基团, 致咪唑环碱性降低; 布立马胺的pKa(H) = 7.25, 说明它的侧链为推电子基团, 致咪唑环碱性增高, 提示环上不同的取代基影响咪唑环的电荷密度, 改变了环上氮原子的碱性。研究表明, R为拉电子基团时, 以图 1A中a的结构占优; R为推电子基团时, 以图 1A中b的结构占优 (Charton M. J Org Chem, 1965, 30: 3346)。
进一步分析, 在生理条件下组胺被质子化程度只有3% (图 1A中c, R=CH2CH2NH2), 80%的存在形式为a, 布立马胺则主要以b的形式存在, 这可能是布立马胺与H2受体结合强度较弱的原因。显然, 咪唑环的高电荷密度是不利的。
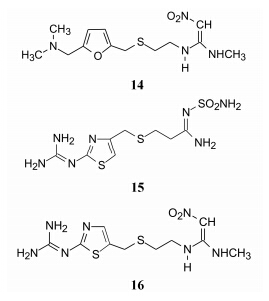
为了降低咪唑环的电荷密度, 在不改变布立马胺分子的形状和长度的前提下, 变侧链为拉电子性质, 为此将侧链中的-CH2-用二价电子等排体-S-替换, 得到化合物8, 称作硫丁咪胺 (thiaburimamide), 由于硫的电负性强于饱和碳, 提高了拮抗活性。进而在8的咪唑环4位引入甲基以有利于形成a的形式, 并将侧链的第2个碳原子用硫置换, 得到甲硫米特 (9, metiamide), 抑制胃酸分泌的强度高于硫丁咪胺3~4倍, 曾进行临床试验治疗胃和十二指肠溃疡, 疗效明确显著, 但少数患者发生粒细胞减少的不良反应 (Thjodleifsson B and Wormsley KG.Gut, 1975, 16: 501)。
改造甲硫米特的结构以消除不良反应, 将硫脲作电子等排变换: 若硫脲换成脲基 (10), 活性显著下降; 变换成胍基 (11), 活性低于甲硫米特20倍, 可能是碱性过强而不利于受体结合, 也不利于药代性质, 为了降低胍基的碱性, N原子上连接拉电子基团如硝基 (12) 或氰基 (13), 对H2受体的拮抗作用与甲硫米特相当, 尤以13为佳, 13有较强的H2拮抗作用及优良的药代和安全性, 作为新的候选药物进入临床研究, 这就是西咪替丁 (cimetidine) (Ganellin R. J Med Chem, 1981, 24: 913; Durant GJ, et al. J Med Chem, 1977, 20: 901)。
西咪替丁又称甲氰咪胍, 经临床研究成为第一个上市的H2受体拮抗剂, 治疗消化道溃疡。

西咪替丁的上市给患者带来福音, 也为SK&F公司带来巨大效益, 成为一个重磅式药物。其他公司的跟踪研发, 相继上市的药物优于西咪替丁 (在当时后继跟踪研发的药物往往优于首创药物是由于首创药物未得到充分优化)。
新研发的药物可以没有组胺的咪唑环, 表明模拟组胺的咪唑环并非H2受体拮抗剂所必须。例如, 以二甲胺甲基呋喃替换甲基咪唑, 氰基胍用硝基亚乙叉二胺代替, 得到的雷尼替丁 (14, ranitidine), 活性强度是西咪替丁的10倍, 与细胞色素P450的作用只是西咪替丁的10%, 口服剂量和每日服用次数减少, 而且对中枢、肾脏和性功能的不良反应也低于西咪替丁, 显示雷尼替丁明显优于西咪替丁。
另一药物是用胍基取代的噻唑环替换西咪替丁的咪唑片段, 用氨磺酰脒代替氰基胍, 即法莫替丁 (15, famotidine), 抑制胃酸分泌作用强于西咪替丁50倍, 作用时间长1.5倍, 并且消除了抗雄激素作用。
尼扎替丁 (16, nizatidine) 是将法莫替丁的噻唑片段与雷尼替丁的侧链相连接, 为按照拼合原理设计的最直观的实例, 在药效、药代和安全性方面尼扎替丁也有许多优点。
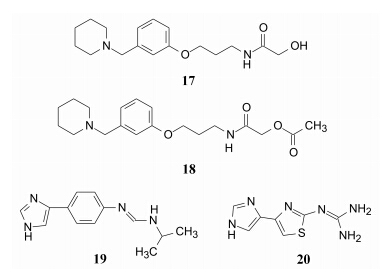
上述后继研发的H2受体拮抗剂在化学结构上 都是与西咪替丁对应性很强的电子等排物, 下面叙 述的药物也是强效的H2拮抗剂, 但结构变化较大。罗沙替丁 (17, roxatidine) 的化学结构, 若以雷尼替丁作参比物, 它的苯环相当于呋喃环, 哌啶甲基对应于二甲胺甲基, 侧链则差异较大, 羟甲酰胺丙氧链 代替了末端成平面结构的碱性片段。哌法替丁 (18, pifatidine) 是罗沙替丁的乙酰化物, 二者都已临床应用, 作用强于西咪替丁。
这些替丁类药物, 结构中都含有一定长度的柔性的亲脂侧链, Donetti等用苯环替换亲脂链, 经骨架迁越研发出新结构类型的咪芬替丁 (19, mifentidine) (Donetti A, et al. J Med Chem, 1989, 32: 957), 抑制胃酸分泌的活性强于西咪替丁30倍。另一刚性更强的药物唑替丁 (20, zaltidine) (Lipinski CA, et al. J Med Chem, 1986, 29: 2154) 的药效和药代性质也优于西咪替丁, 分子中咪唑环、噻唑环和胍基由单键连接, 形成共轭体系, 已经完全不同于首创药物西咪替丁所具有的结构特征, 说明药物结构的变换引起分子构象的变化和药效团空间分布的变动, 但仍可实现与H2受 体的结合。