 2015, Vol. 50
2015, Vol. 50



核糖体蛋白S6激酶 (ribosomal protein S6 kinase) 家族由p90RSK和p70S6K两种亚族组成,因其能调控40S核糖体亚单位S6蛋白的磷酸化而得名,人体中,主要受p70S6K调控。p70S6K包括S6K1和S6K2两个亚型,其序列一致性达到83%。其中S6K1是雷帕霉素靶蛋白 (mammalian target of rapamycin,mTOR) 下游的一个重要效应器,目前对S6K2生物功能的研究还不深入。正常生理条件下,细胞膜外的有丝分裂原、胰岛素、细胞应激、氨基酸等激活细胞内mTOR通路,进而通过S6K1调控细胞转录、翻译、血管新生以及凋亡等生物学过程 (图 1)[1]。

|
图1 mTOR/p70S6K信号通路[1] |
肿瘤的发生一般是由于细胞的生长、凋亡等失去正常调控而造成的。研究表明,mTOR在肿瘤发展过程中起关键作用,例如前列腺癌、胃癌等肿瘤的生长过程中都发现mTOR过度表达及突变[2, 3]。表皮生长因子、RAS以及PI3K等一系列致癌信号分子都能上调mTOR的表达,从而促进肿瘤细胞的生长[4, 5, 6]。因此,p70S6K成为一个潜在的药物靶点,寻找p70S6K抑制剂成为肿瘤治疗的一个新策略。
在肿瘤早期,氨基酸及葡萄糖转移蛋白水平上调,激活mTOR/p70S6K信号通路,促进核糖体生物合成、细胞生长以及抑制细胞的自我吞噬,使肿瘤进一步恶化。p70S6K通过调节其下游效应器,介导肿瘤发展过程的多个重要环节,成为致癌的一个潜在因素。
肿瘤最明显的特征就是细胞生长失调,S6K1通过S6蛋白介导细胞的生长过程。基因敲除S6K1的小鼠,其器官出现萎缩,体型较对照组明显变小。体外和体内实验均证明,多种肿瘤细胞的S6K1基因敲除后,其生长都受到抑制[7]。
血管新生是肿瘤生长的一个重要过程,其中 血管内皮生长因子 (VEGF) 起关键性作用,S6K1通 过HIF-1α介导VEGF的表达,当用PI3K抑制剂 (LY294002) 间接抑制S6K1后,VEGF活性下调,从而抑制肿瘤发展进程[8]。
诱导细胞凋亡是肿瘤治疗的一种策略,B细胞淋巴瘤家族蛋白对细胞凋亡的调控起主要作用,其中属于该家族的BAD蛋白能够促进细胞凋亡。激活S6K1后,会抑制BAD蛋白的活性,从而阻碍细胞 凋亡。这种调控行为导致S6K1诱导卵巢癌细胞对紫杉醇产生耐药性[9]。在小细胞肺癌中,S6K2会增强纤维母细胞生长因子2 (FGF-2) 诱导的耐药性。但敲除S6K2基因后,抑制Akt活性,同时诱导乳腺癌细胞 凋亡[10]。
PI3K抑制剂LY-294002通过间接抑制S6K1的活性,诱发卵巢癌细胞由G1期转入S期,进而抑制卵巢癌细胞的增殖[11]。Yamnik等[12]发现,S6K1高表达后,上调雌激素受体α (ERα) 的转录活性,从而促进乳腺癌细胞增殖。下调S6K1的表达后,对正常细胞无影响,但可以抑制乳腺癌细胞增殖。
同时,体外研究发现,p70S6K能负反馈调节Akt的活性。当p70S6K激活后,会磷酸化IRS-1磷酸酪氨酸结合区 (PTB) 位点Ser 302,从而抑制IRS-1与胰岛素受体相结合。此外,p70S6K还会通过一种未知机制下调IRS-1的表达。最新研究表明,S6K1能够磷酸化mTORC2复合物中Rictor的位点Thr 1135,从而介导mTORC2-Akt通路抑制Akt的活性[13],最终反馈性抑制mTOR/p70S6K信号通路[14]。
在哺乳动物中,p70S6K均含有几个重要的调控区域: ① 含有雷帕霉素靶标的酸性N端; ② 含有T-loop的激酶区域; ③ 含有转角模体 (TM) 和疏水模体 (HM) 的连接区域; ④ 假底物自磷酸化区域的C端[15]。S6K1整体拓扑结构与AGC家族其他激酶相似 (图 2): N端和C端区域分别由β折叠及α螺旋构成,其中,αC螺旋和活化环分别位于N端残基130~140区及C端243~255区,在非磷酸化条件下,其处于失调状态。切除C端自磷酸化区域后,S6K1活性不受影响,但连接区域位点Thr 389和激酶区域位点Thr 229发生突变,会导致S6K1活性降低甚至失活,说明S6K1的活性与这两个位点的氨基酸残基密切相关[16]。S6K1的ATP结合口袋位于C端和N端之间的疏水穴,这个口袋中的关键残基与AGC家族其他激酶相比,具有一定的特异性。

|
图2 A: S6K1晶体结构; B: S6K1和Akt晶体结构叠合 |
S6K1活化机制有多种假说,目前公认的是,ERK、p38、CDC2以及JNK1等丝氨酸激酶先磷酸化C端自磷酸区域的Ser 411、Ser 418、Ser 421、Ser 424,使失活状态下的S6K1发生构象转换,暴露出连接区域和激酶区域,然后mTORC1磷酸化连接区域HM位置的Thr 389,使激酶区域产生疏水口袋,诱导PDK1作用片段 (PIF) 蛋白与S6K1结合,从而为PDK1产生了一个停泊位置,使其磷酸化激酶区的Thr 229,最后完全激活S6K1[17]。
雷帕霉素及其类似物能间接抑制p70S6K活性,近年来,临床上通过不同给药方案治疗肿瘤[18, 19, 20],表现了令人鼓舞的结果。对于结节性硬块症,雷帕霉素的治疗效果很好[21],这说明以p70S6K为靶点,用于肿瘤治疗是合理有效的。雷帕霉素及其类似物的 水溶性不好,而且临床使用过程中出现了耐药性及副作用[22],因此,人们开始研究p70S6K抑制剂,更有效的治疗肿瘤。由于p70S6K对Akt存在负反馈 调节,因此p70S6K抑制剂的研究有两种方向: 针对p70S6K的单靶点抑制剂和同时作用于p70S6K和Akt的双靶点抑制剂[23]。
通过与ATP竞争性结合来阻断酶的催化过程,从而阻断细胞信号通路的传导来达到治疗目的,是以激酶为靶点进行药物设计的一个基本思路。目前研究中的p70S6K抑制剂大多属于ATP竞争性抑制剂,根据化合物结构的不同,将其分为嘧啶类、双吲哚马来酰亚胺类、苯并咪唑类、二硫杂环类、吡唑并嘧啶类衍生物等。
PF-4708671 (1) 是辉瑞公司研发的哌嗪基嘧啶类化合物 (图 3),它是S6K1的第一个特异性抑制剂,其抑制常数Ki及IC50分别为20和160 nmol·L-1,对S6K2抑制作用并不显著[24]。Wang等[25]发现PF-4708671作用于S6K1的ATP结合区域: 咪唑环的氨基以及嘧啶环的3位N分别与Asp 213、Glu 150、Leu 152形成氢键; 三氟甲基 与Gly 80产生卤键作用; 嘧啶环上的乙基与Val 82还存在疏水作用力。PF-4708671与S6K1结合后,使原本失调的αC螺旋变得稳定,从而迅速诱导S6K1活化位点 (Thr 389及Thr 229) 的磷酸化,抑制S6K1活性。

|
图3嘧啶类衍生物 |
LY2584702 (2) (图 3) 是礼来公司研发的嘧啶并吡唑类化合物,能有效的抑制S6K1,IC50值为4 nmol·L-1。其口服给药用于治疗实体瘤已经完成一期临床试验,结果表明其并不能有效的治疗实体瘤,不过受试者没有产生与雷帕霉素相类似的高总胆固醇副作用[26]。Hollebecque等[27]将LY2584702与依维莫司或埃罗替尼联合给药用于治疗实体瘤,但试验过程中,受试者出现了紧急凝血障碍,因此目前还没决定是否进入二期临床。
十字孢碱 (3) 能竞争性抑制丝氨酸/苏氨酸及酪氨酸激酶的ATP结合位点,但其选择性不高。Morreale等[28]在开发PKC选择性激酶抑制剂过程中,发现化合物Ro31-8220 (4)、GF109203X (5) 是PKC与p70S6K双靶点抑制剂,对Ro31-8220进行结构改造,得到p70S6K单靶点抑制剂Ro 31-6045 (6),该化合物活性虽然不高,但对设计开发p70S6K选择性抑制剂,具有一定参考价值 (图 4)[29]。

|
图4双吲哚马来酰亚胺类衍生物 |
研究表明,具有3-氨基- 1,2,5-噁二唑母核的化合物能够抑制丝氨酸/苏氨酸激酶活性。Bandarage研究组[30]以PKAα (PDB: 1jbp) 为模板,同源模建了S6K1的ATP结合口袋,通过对接发现氨基以及噁二唑2位N分别与Glu 173羰基及Leu 175氨基形成氢键结合,并且该口袋还具有A、B 两个一大一小的结合腔 (图 5)。

|
图5苯并咪唑类衍生物模拟结合位点 |
Bandarage等首先对苯并咪唑环的N-1位进行结构修饰 (表 1),结果表明,体积较大的取代基能显著提高S6K1抑制活性。但随着取代基的极性增大,与S6K1的结合能力反而越弱; N-1苯环邻位取代的活 性要明显高于间、对位取代,芳香型取代基选择性要高于脂肪型取代基,原因可能是AGC家族的其他激酶与A腔相对应的氨基酸的残基为Leu,因此形成的口袋难以容纳芳香型取代基,故活性不高。这种特有的结构特点有利于设计S6K1特异性抑制剂。
|
|
表1 苯并咪唑类衍生物N-1位取代构效关系 |
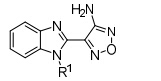
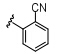
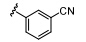
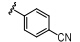
以活性最好的化合物9为基础,对苯并咪唑的5位进行结构优化,使其与B腔结合 (表 2),结果表明小体积取代基对S6K1抑制活性增强,与前期计算机辅助药物设计的结合模型基本一致,表明与B腔的结合能够提高化合物对S6K1的抑制作用。
|
|
表2 苯并咪唑类衍生物5位取代构效关系 |
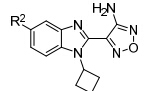
Couty等[31]对S6K1的活化区域用AlphaScreenTM技术在类先导化合物库中筛选,同样发现3-氨基-1,2,5-噁二唑母核对S6K1具有抑制活性。将化合物13与十字孢碱-S6K1晶体结构 (PDB: 3A60) 叠合,发现该类化合物为S6K1的ATP竞争性抑制剂。前期 研究表明,氮杂苯并咪唑对S6K1具有很强的亲和力。Couty等用氮杂苯并咪唑代替苯并咪唑环,并且对氮杂苯并咪唑6位进行结构修饰 (表 3),研究其对S6K1抑制活性。表明小基团取代保持了一定的抑制活性和相对高的配体亲和力。将化合物18、22与PKA-S6K1嵌合体 叠合发现,与苯并咪唑5位取代基相类似,氮杂苯并咪唑6位取代基可与一个小疏水腔作用。
|
|
表3 苯并咪唑类衍生物6位取代构效关系 |
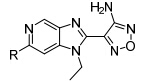
在对胰岛素受体耐受性研究中,Bae等[32]发现奥替普拉 (23) 能够抑制S6K1活性 (图 6),因此他们便以二硫杂环类衍生物作为先导化合物,研究以1,2-二硫杂环戊二烯-3-硫酮为母核的化合物对S6K1的抑制活性。通过细胞实验发现,化合物CJ11766 (24)、CJ11788 (25)、CJ11792 (26)、CJ11842 (27) 以及CJ12064 (28) 对S6K1有显著的抑制作用。其中奥替普拉通过抑制S6K1活性来治疗非酒精脂肪肝研究正处于三期临床试验。

|
图61,2-二硫杂环类衍生物 |
化合物29是p38抑制剂,Ye研究组[33]以其为先导化合物,进行结构优化 (表 4),结果表明: 杂环芳醚中的N原子在3位时,具有抑制活性,对其构象进行限制,或用取代苯替代吡啶环,抑制活性都会降低甚至消失,引入苯并咪唑环,活性明显提高。Ye等认为,在这个位置,需要一个构象限制的氢键受体。
|
|
表4 尿素类衍生物构效关系 |
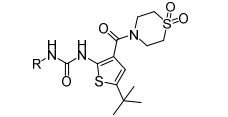
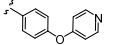
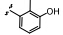
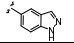
在化合物32的基础上,对噻吩环继续结构优化,结果表明: 噻吩的5位以疏水基团取代活性最好,当叔丁基被功能相类似的甲基 (IC50 = 223 nmol·L-1)、乙基 (IC50 = 62 nmol·L-1)、异丙基(IC50 = 29 nmol·L-1)、环丙基 (IC50 = 70 nmol·L-1) 替换时,活性均下降,苯环取代时,活性基本消失。噻吩3位取代基的替换不影响化合物的抑制活性,但可以改变化合物的理化性质 (化合物33)。
综合该系列化合物的性质,可以推测出该类抑制剂与S6K1相结合的关键药效团因素 (图 7): 能与铰链区形成氢键的氢键受体; 与疏水口袋相结合的疏水基团以及处于溶剂暴露区域的极性基团。

|
图7尿素类似物模拟结合图 |
Bussenius等[34]通过高通量筛选,发现化合物34对p70S6K具有抑制活性 (图 8),对其进行结构优化发现: 苯环引入取代基的活性比未取代好; 嘧啶环上的氨基和硝基可以成环但不能替换,环合后的化合物36虽然具有酶活性,但对A549细胞的抑制活性并不理想。对化合物36进行构效关系讨论: 3位以疏水性基团取代活性最好; 苯环间位取代不影响活性,但可以改善该类化合物的水溶性,从而提高其成药性。对大鼠经口给药,发现化合物39的生物利用度得到了极大改善,具有一定的研究前景。

|
图8吡唑并嘧啶类衍生物 |
Oh等[35]发现氨基葡萄糖对MDA- MB-231乳腺癌细胞以及DU145前列腺癌细胞的增殖具有抑制作用。研究表明,氨基葡萄糖是p70S6K非ATP竞争性抑制剂,能够抑制p70S6K位点Thr 389的磷酸化,从而具有抗肿瘤的作用。这类化合物能够与p70S6K特异性结合,因此,可以为设计p70S6K特异性抑制剂结构提供参考依据,提高抑制剂的选择性,同时降低耐药性。
由于合成药物普遍存在不良副作用,人们现在越来越关注天然产物在临床中的应用,目前已经发现一些天然产物可以下调肿瘤细胞中S6K的表达,或是抑制肿瘤细胞增殖过程中p70S6K的活性[36]。如表 5所示[37, 38, 39, 40, 41, 42, 43, 44, 45, 46]。
非瑟酮 (40,图 9) 是从水果及蔬菜中提取出的活性小分子,能抑制肿瘤细胞增殖,Syed等[47]认为这是因为非瑟酮能够抑制p70S6K的活性,通过对接发现,其是ATP竞争性抑制剂。据此,他们开始研究非瑟酮对黑色素瘤的治疗作用。

|
图9非瑟酮结合示意图 |
Zhang等[48]分析S6K1抑制剂结构特征后,借助计算机辅助药物设计,构建了S6K1抑制剂的药效团模型 (图 10),即: 1个氢键受体、1个氢键供体、1个疏水结合特征以及1个芳香环特征。在此药效团模型的基础上,筛选出一些对S6K1有潜在抑制作用的化合物,把打分最高的化合物AG227与S6K1晶体结构 (PDB: 3A60) 对接,发现该化合物能够与S6K1的ATP口袋关键残基Leu175、Glu179、Met225很好的结合,其中链状丁酸处于溶剂暴露区域,这与尿素类衍生物相类似。
|
|
表5 具有p70S6K抑制活性的天然产物 |

|
图10A: S6K1药效团因素; B: AG227结构与S6K1药效团叠合 |
基于药效团技术来进行全新药物结构设计和先导化合物优化,可以提高药物研发的成功率,降低研发成本。通过对p70S6K抑制剂药效团的研究,人们能够发现母核结构更新颖、活性更好的化合物。
雷帕霉素及其类似物 (CCI-779,RAD-001等) 的临床前研究发现,阻滞mTORC1的下游信号传导时,
可以明显观察到Akt活性上调[49],p70S6K抑制剂和mTOR抑制剂联合用药,对胶质细胞瘤大鼠模型的治疗效果比单一给药形式要好。这些现象使人们开始关注p70S6K和Akt双靶点抑制剂的研究,从而避免这种负反馈调节对p70S6K单靶点抑制剂治疗效果的影响。
AT7876 (41) 对Akt的3种亚型都具有一定的抑制作用,其中对Akt2抑制活性最好。AT7876的吡唑环能与Akt2铰链区Glu 230及Ala 232形成氢键,增强对Akt的抑制作用。AT7876通过口服给药能够有效的抑制肿瘤细胞的增殖,而且比单靶点抑制剂更有效[50]。
AZD8055 (42) 是由阿斯利康研发的吡啶并吡嗪类化合物 (图 11),其能抑制Akt通路的mTORC2位点 (Ser 473) 的磷酸化,从而阻断Akt通路,在某些细胞株中,AZD8055还能抑制Akt的Thr 308位点磷酸化。对p70S6K的抑制作用为剂量依赖型,在MCF-7和HEK细胞中,AZD8055对p70S6K的抑制率分别为70% 及80%,还能有效的抑制U87MG、A549及H838细胞增殖 (IC50分别为53、50、20 nmol·L-1)。AZD8055用于治疗神经胶质瘤、实体瘤、恶性肿瘤已经完成一期临床试验,但目前还没有公布数据结果[51]。

|
图11AT7876和AZD8055结构 |
惠氏公司[52]对苯并2,7-二氮杂萘类衍生物研究发现,化合物结构上的微小差别都会改变化合物对这 两个激酶的选择性。如图 12所示,化合物43是双靶点抑制剂,在苯环的邻、对位引入羟基或氯取代后 (44和45),依旧是双靶点抑制剂,但同时在苯环间、对位引入氯取代则导致Akt抑制活性的消失。辉瑞和诺华公司对苯并吡啶类衍生物及具有1H-咪唑喹啉母核的化合物研究过程中也发现类似的情况。

|
图122,7-二氮杂萘类衍生物结构 |
Rice等[53]在化合物39的基础上,发现吡唑并嘧啶类化合物也能抑制Akt1的活性,继续对吡唑并嘧啶的3位及氨基侧链进行结构优化,提高了对Akt1的抑制活性 (图 13)。将Akt抑制剂 (50) 及化合物37同时与Akt晶体(PDB: 1O6L) 对接,发现化合物37苯环的5位与Akt结合口袋可以疏水性结合。因此,Rice等在苯环的5位引入疏水基团 (48和49),对p70S6K及Akt均有抑制作用。动物实验表明化合物49的生物利用度较高,狗及猴对49的口服生物利用度为58% 和77%。但由于低的药物暴露量,化合物49终止了用于实体瘤治疗的一期临床试验。

|
图13吡唑并嘧啶类双靶点抑制剂 |
随着分子生物学研究的深入发展,人们发现许多疾病的发生都是多因素的,尤其是在肿瘤的发生发展中,信号分子存在不同信号传导途径,这些都致 使疾病治疗变得越来越复杂,因此,针对疾病成因的多靶点作用药物将有利于提高药物疗效,降低副作用,达到药物设计的理想目的[54]。
目前,S6K1特异性抑制剂已经开始用于卵巢癌以及其他肿瘤的治疗研究。尽管所有激酶的催化区域都具有高度的保守性,但不同亚型在三维晶体结构上还是存在一定的差异。随着对p70S6K激酶晶体结构及生物学功能逐渐了解,人们将会研究出更多具有新型骨架的p70S6K特异性抑制剂,从而避免传统药物治疗过程中所出现的副作用及耐药性。此外,进一步了解p70S6K信号通路,开发p70S6K和Akt双靶点抑制剂,解决p70S6K抑制剂对同源蛋白选择性差的难题,且达到联合用药的效果,这点在抗肿瘤药物研究开发中具有光明的前景。
| [1] | Dann SG, Selvaraj A, Thomas G, et al. mTOR complex1- S6K1 signaling: at the crossroads of obesity, diabetes and cancer [J]. Trends Mol Med, 2007, 13: 252-259. |
| [2] | Sutherland SIM, Pe Benito R, Henshall SM, et al. Expression of phosphorylated-mTOR during the development of prostate cancer [J]. Prostate, 2014, 74: 1231-1239. |
| [3] | Matsuoka T, Yashiro M. The role of PI3K/Akt/mTOR signaling in gastric carcinoma [J]. Cancers, 2014, 6: 1441- 1463. |
| [4] | Zoncu R, Efeyan A, Sabatini DM, et al. mTOR: from growth signal integration to cancer, diabetes and ageing [J]. Nat Rev Mol Cell Biol, 2011, 12: 21-35. |
| [5] | Vega F, Medeiros LJ, Leventaki V, et al. Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma [J]. Cancer Res, 2006, 66: 6589-6597. |
| [6] | Riemenschneider MJ, Betensky RA, Pasedag SM, et al. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling [J]. Cancer Res, 2006, 66: 5618-5623. |
| [7] | Bian CX, Shi Z, Meng Q, et al. p70S6K1 regulation of angiogenesis through VEGF and HIF-1alpha expression [J]. Biochem Biophys Res Commun, 2010, 398: 395-399. |
| [8] | Skinner HD, Zheng JZ, Fang J, et al. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia- inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling [J]. J Biol Chem, 2004, 279: 45643-45651. |
| [9] | Foster H, Coley HM, Goumenou A, et al. Differential expression of mTOR signalling components in drug resistance in ovarian cancer [J]. Anticancer Res, 2010, 30: 3529-3534. |
| [10] | Sridharan S, Basu A. S6 kinase 2 promotes breast cancer cell survival via Akt [J]. Cancer Res, 2011, 71: 2590-2599. |
| [11] | Gao N, Flynn DC, Zhang Z, et al. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/ mTOR/p70S6K1 signaling in human ovarian cancer cells [J]. Am J Physiol Cell Physiol, 2004, 287: C281-C291. |
| [12] | Yamnik RL, Digilova A, Davis DC, et al. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation [J]. J Biol Chem, 2009, 284: 6361-6369. |
| [13] | Sparks C, Guertin D. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy [J]. Oncogene, 2010, 29: 3733-3744. |
| [14] | Harrington LS, Findlay GM, Gray A, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins [J]. J Cell Biol, 2004, 166: 213-223. |
| [15] | Sunami T, Byrne N, Diehl RE, et al. Structural basis of human p70 ribosomal S6 kinase-1 regulation by activation loop phosphorylation [J]. J Biol Chem, 2010, 285: 4587- 4594. |
| [16] | Keshwani MM, von Daake S, Newton AC, et al. Hydrophobic motif phosphorylation is not required for activation loop phosphorylation of p70 ribosomal protein S6 kinase 1 (S6K1) [J]. J Biol Chem, 2011, 286: 23552-23558. |
| [17] | Zhang J, Gao Z, Ye J. Phosphorylation and degradation of S6K1 (p70S6K1) in response to persistent JNK1 activation [J]. Biochim Biophys Acta, 2013, 1832: 1980-1988. |
| [18] | Yuan R, Kay A, Berg WJ, et al. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy [J]. J Hematol Oncol, 2009, 2: 45. |
| [19] | Joka M, Boeck S, Zech CJ, et al. Combination of antiangiogenic therapy using the mTOR-inhibitor everolimus and low- dose chemotherapy for locally advanced and/or metastatic pancreatic cancer: a dose-finding study [J]. Anticancer Drugs, 2014, 25: 1095-1101. |
| [20] | Ng VC, Johnson JJ, Cuellar S. Targeting the mammalian target of rapamycin pathway with everolimus: implications for the management of metastatic breast cancer [J]. J Oncol Pharm Pract, 2014, pii: 1078155214540732. |
| [21] | Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex [J]. Ann Neurol, 2006, 59: 490-498. |
| [22] | Rodriguez-Pascual J, Cheng E, Maroto P, et al. Emergent toxicities associated with the use of mTOR inhibitors in patients with advanced renal carcinoma [J]. Anti-Cancer Drugs, 2010, 21: 478-486. |
| [23] | Fenton TR, Gout IT. Functions and regulation of the 70 kDa ribosomal S6 kinases [J]. Int J Biochem Cell Biol, 2011, 43: 47-59. |
| [24] | Pearce L, Alton G, Richter D, et al. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) [J]. Biochem J, 2010, 431: 245- 255. |
| [25] | Wang J, Zhong C, Wang F, et al. Crystal structures of S6K1 provide insights into the regulation mechanism of S6K1 by the hydrophobic motif [J]. Biochem J, 2013, 454: 39-47. |
| [26] | Tolcher A, Goldman J, Patnaik A, et al. A phase I trial of LY2584702 tosylate, a p70 S6 kinase inhibitor, in patients with advanced solid tumours [J]. Eur J Cancer, 2014, 50: 867-875. |
| [27] | Hollebecque A, Houédé N, Cohen EEW, et al. A phase Ib trial of LY2584702 tosylate, a p70 S6 inhibitor, in combination with erlotinib or everolimus in patients with solid tumours [J]. Eur J Cancer, 2014, 50: 876-884. |
| [28] | Morreale A, Mallon B, Beale G, et al. Ro31-8220 inhibits protein kinase C to block the phorbol ester-stimulated release of choline-and ethanolamine-metabolites from C6 glioma cells: p70 S6 kinase and MAPKAP kinase-1β do not function downstream of PKC in activating PLD [J]. FEBS Lett, 1997, 417: 38-42. |
| [29] | Marmy-Conus N, Hannan KM, Pearson RB. Ro 31-6045, the inactive analogue of the protein kinase C inhibitor Ro 31-8220, blocks in vivo activation of p70S6K/p85S6K: implications for the analysis of S6K signalling [J]. FEBS Lett, 2002, 519: 135-140. |
| [30] | Bandarage U, Hare B, Parsons J, et al. 4-(Benzimidazol-2-yl)- 1, 2, 5-oxadiazol-3-ylamine derivatives: potent and selective p70S6 kinase inhibitors [J]. Bioorg Med Chem Lett, 2009, 19: 5191-5194. |
| [31] | Couty S, Westwood IM, Kalusa A, et al. The discovery of potent ribosomal S6 kinase inhibitors by high-throughput screening and structure-guided drug design [J]. Oncotarget, 2013, 4: 1647-1661. |
| [32] | Bae EJ, Yang YM, Kim JW, et al. Identification of a novel class of dithiolethiones that prevent hepatic insulin resistance via the adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway [J]. Hepatology, 2007, 46: 730-739. |
| [33] | Ye P, Kuhn C, Juan M, et al. Potent and selective thiophene urea-templated inhibitors of S6K [J]. Bioorg Med Chem Lett, 2011, 21: 849-852. |
| [34] | Bussenius J, Anand NK, Blazey CM, et al. Design and evaluation of a series of pyrazolopyrimidines as p70S6K inhibitors [J]. Bioorg Med Chem Lett, 2012, 22: 2283-2286. |
| [35] | Oh HJ, Lee JS, Song DK, et al. D-glucosamine inhibits proliferation of human cancer cells through inhibition of p70S6K [J]. Biochem Biophys Res Commun, 2007, 360: 840-845. |
| [36] | Ip CK, Wong AS. Exploiting p70 S6 kinase as a target for ovarian cancer [J]. Expert Opin Ther Targets, 2012, 16: 619- 630. |
| [37] | Jeong JH, Jeong YJ, Cho HJ, et al. Ascochlorin inhibits growth factor-induced HIF-1α activation and tumor- angiogenesis through the suppression of EGFR/ERK/p70S6K signaling pathway in human cervical carcinoma cells [J]. J Cell Biochem, 2012, 113: 1302-1313. |
| [38] | Johnson SM, Gulhati P, Arrieta I, et al. Curcumin inhibits proliferation of colorectal carcinoma by modulating Akt/ mTOR signaling [J]. Anticancer Res, 2009, 29: 3185-3190. |
| [39] | Alkhalaf M. Resveratrol-induced apoptosis is associated with activation of p53 and inhibition of protein translation in T47D human breast cancer cells [J]. Pharmacology, 2007, 80: 134-143. |
| [40] | He X, Wang Y, Zhu J, et al. Resveratrol enhances the anti- tumor activity of the mTOR inhibitor rapamycin in multiple breast cancer cell lines mainly by suppressing rapamycin- induced AKT signaling [J]. Cancer Lett, 2011, 301: 168- 176. |
| [41] | Fang J, Xia C, Cao Z, et al. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways [J]. FASEB J, 2005, 19: 342-353. |
| [42] | Kuo TC, Chiang PC, Yu CC, et al. A unique P-glycoprotein interacting agent displays anticancer activity against hepatocellular carcinoma through inhibition of GRP78 and mTOR pathways [J]. Biochem Pharmacol, 2011, 81: 1136-1144. |
| [43] | Sun YW, Huang WJ, Hsiao CJ, et al. Methoxychalcone induces cell-cycle arrest and apoptosis in human hormone- resistant prostate cancer cells through PI 3-kinase-independent inhibition of mTOR pathways [J]. Prostate, 2010, 70: 1295- 1306. |
| [44] | Zhang B, Huang H, Xie J, et al. Cucurmosin induces apoptosis of BxPC-3 human pancreatic cancer cells via inactivation of the EGFR signaling pathway [J]. Oncol Rep, 2012, 27: 891. |
| [45] | Yu CC, Chiang PC, Lu PH, et al. Antroquinonol, a natural ubiquinone derivative, induces a cross talk between apoptosis, autophagy and senescence in human pancreatic carcinoma cells [J]. J Nutr Biochem, 2012, 23: 900-907. |
| [46] | Lee MS, Cha EY, Sul JY, et al. Chrysophanic acid blocks proliferation of colon cancer cells by inhibiting EGFR/mTOR pathway [J]. Phytother Res, 2011, 25: 833-837. |
| [47] | Syed DN, Chamcheu JC, Khan MI, et al. Fisetin inhibits human melanoma cell growth through direct binding to p70S6K and mTOR: findings from 3-D melanoma skin equivalents and computational modeling [J]. Biochem Pharmacol, 2014, 89: 349-360. |
| [48] | Zhang H, Xiang ML, Liang JY, et al. Combination of pharmacophore hypothesis, genetic function approximation model, and molecular docking to identify novel inhibitors of S6K1 [J]. Mol Divers, 2013, 17: 767-772. |
| [49] | O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt [J]. Cancer Res, 2006, 66: 1500-1508. |
| [50] | Grimshaw KM, Hunter LJK, Yap TA, et al. AT7867 is a potent and oral inhibitor of AKT and p70 S6 kinase that induces pharmacodynamic changes and inhibits human tumor xenograft growth [J]. Mol Cancer Ther, 2010, 9: 1100-1110. |
| [51] | Chresta CM, Davies BR, Hickson I, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity [J]. Cancer Res, 2010, 70: 288-298. |
| [52] | Kim KH, Wissner A, Floyd MB, et al. Benzo[c][2,7] naphthyridines as inhibitors of PDK-1 [J]. Bioorg Med Chem Lett, 2009, 19: 5225-5228. |
| [53] | Rice KD, Kim MH, Bussenius J, et al. Pyrazolopyrimidines as dual Akt/p70S6K inhibitors [J]. Bioorg Med Chem Lett, 2012, 22: 2693-2697. |
| [54] | Jiang FC. The multi-target drugs and their design [J]. Acta Pharm Sin (药学学报), 2009, 44: 282-287. |