 2015, Vol. 50
2015, Vol. 50

依折麦布是通过抑制肠道吸收胆固醇的作用机制降低体内胆固醇的首创性药物。
1 作用靶标人体内的胆固醇来源有两个途径: 自身合成和膳食摄取。体内近三分之二的胆固醇是自身合成的, 由乙酰辅酶A经30多个酶催化的生化反应生成; 其余部分来自于膳食, 经肠吸收进入肝脏。负责胆固醇吸收的一个靶标, 是酰化辅酶A胆固醇酰基转移酶 (acyl-coenzyme A cholesterol acyltransferase, ACAT)。ACAT是微粒体酶, 负责将游离胆固醇转变为脂肪酸胆固醇酯, 抑制ACAT可以降低胆固醇的肠内吸收, 减少胆固醇从膳食的摄取量, 降低血浆中胆固醇水平。此外, ACAT来源的胆固醇酯还参与动脉斑块的形成, 所以ACAT抑制剂可减缓动脉硬化的进程。
2 苗头化合物和向先导物的演化先灵葆雅公司经随机筛选发现化合物1对体外ACAT有一定抑制作用, 作为初步苗头, 通过结构变换和构效关系研究, 得到化合物2, 活性有所提高, 2对ACAT抑制活性IC50 = 0.9 μmol·L-1, 大鼠灌胃 50 mg·kg-1, 可降低高胆固醇饲料大鼠肝脏中胆固醇酯80%, 但血浆中的胆固醇水平变化不显著 (Clader JW, et al. J Med Chem, 1995, 38: 1600)。分析苗头化 合物的结构, 有8个可旋转键, 分子过于柔性, 这对提高活性构象体是不利的。在由2演化成先导物 (hit-to-lead) 过程中, 采用了构象限制策略, 用不同的成环方式, 合成了二氢茚胺 (3) 和两种四氢异喹啉 (4和5) 化合物 (Vaccaro W, et al. J Med Chem, 1996, 39: 1704)。

这3类构象限制性化合物仍然具有活性, 其中骨架为二氢茚胺的化合物(3, R=OH) 对ACAT抑制活性显著提高, IC50 = 50 nmol·L-1, 降低肝脏胆固醇酯水平达88%。苗头化合物2的另一种构象限制体是非稠合性杂环, 即以氮杂环丁烷为骨架的分子6 (Duane A, et al. J Med Chem, 1994, 37: 1733), 但6只有微弱的抑制活性。

化合物6的合成是经烯醇酯与亚胺缩合生成β-内酰胺7, 再经还原而生成的。反应中, 缩合的初始产物可发生立体选择性的酰化反应, 生成副产物8。测定中间体7和副产物8的活性, 发现8虽然对ACAT活性较弱 (IC50 = 7 μmol·L-1), 却可以降低体内的肝脏胆固醇酯水平, 而且也降低血浆中胆固醇浓度, 而6和7体内活性很弱 (Clader JW, et al. J Med Chem, 1996, 39: 3684)。反应副产物8的偶然发现, 成为研究转折点。
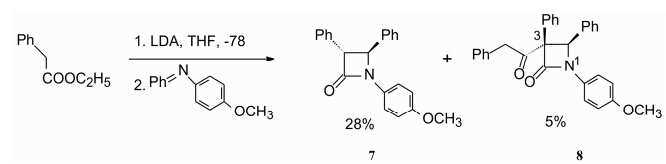
将8作为新的苗头物, 合成了一系列衍生物, 重点考察C3位取代基对活性的影响。结果表明, 芳烷基取代的活性更强而且是必要的, 换成3-烷基取代则失去活性。化合物9(SCH 48461) 虽然体外对ACAT的活性很弱 (IC50 = 26 μmol·L-1), 但体内降胆固醇活性很强, 在灌胃1~2 mg·kg-1剂量下对仓鼠、大鼠、犬和猴都能显著降低肝脏胆固醇酯和血浆中胆固醇水平 (Salisbury BG, et al. Atherosclerosis, 1995, 115: 45)。体外与体内的活性不一致的现象在新药研究中时有发生, 应当以体内活性为准。因而以后的化合物活性都用动物实验进行评价。
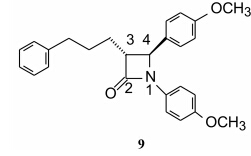
由于9的体内活性高, 将其作为先导化合物, 并以降低体内胆固醇作为药效学指标进行结构优化。为此, 对各个位置进行了系统的研究, 经动物实验评价, 得到结构与活性关系的要点如下: 1位的苯环是必须的, 若被烷基、芳烷基或芳酰基代替, 则失去活性。苯环上有或没有取代基、取代基的性质和在芳环上所处的位置对活性影响较小; 2位的羰基是必要的, 还原成亚甲基失去活性; 3位的芳烷基的构型很重要, C3为R-构型的活性强于S-构型。链的碳原子数对活性的影响次序为3 > 4 > 2, 所以3个碳原子为最佳。
在化合物9的3-位侧链中引入双键, E-构型化合物10的活性 (50 mg·kg-1, 降低胆固醇18%) 明显低于Z-构型化合物11 (50 mg·kg-1, 降低胆固醇95%), 提示顺式双键的活性强于反式构型, 说明3-位侧链上苯环的空间走向和位置不同, 可影响与受体蛋白的结合方式, 活性相差较大。

根据3R和3S的立体化学及其活性, 推测分子 与靶标结合时的构象, 是3-位侧链无论是R或S构 型, 苯环在空间上处于大致相同的结合位置, 如图 1所示。3-位碳原子双取代后由于位阻原因, 使苯环偏离结合位置而活性降低 (Dugar S, et al. J Med Chem, 1995, 38: 4875)。按照这个模型合成的顺式和反式螺环化合物, 应当符合这种构象模型。

|
Figure 1 根据构象模型设计的螺环化合物 |
实际上反式化合物12的降低胆固醇活性强于化合物9, 而顺式螺环化合物13没有活性, 其他螺环 化合物也是反式活性强于顺式。

4位的苯环是必须的, S构型强于R。4-甲氧苯基是重要的药效团特征, 变换到2-位或3-位或换成其他基团均降低活性, 但4-羟基仍有活性, 推论4-羟基是4-甲氧基的活化形式。甲氧基若被不能形成氢键的原子或基团取代, 则失去活性。
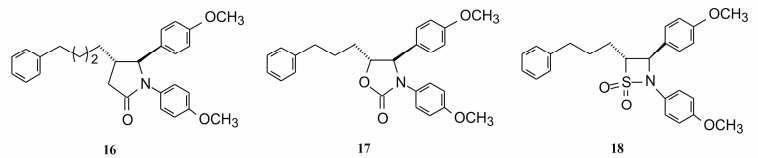
中心环的变换: 中心环扩环成五元γ-内酰胺或其他五元杂环仍有活性, 但活性弱于四元环。例如, 开环化合物14, 氮杂环丁烷15, 失去活性; 五元环γ-内酰胺16、噁唑烷酮17、β-环磺酰胺18等虽然有活性, 但都低于β-内酰胺环。
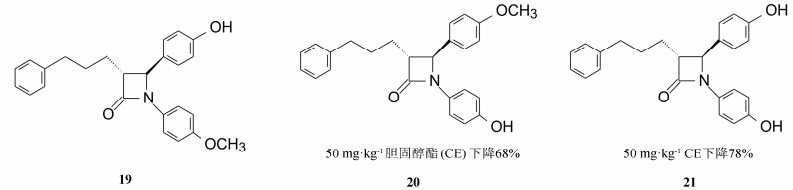
根据上面的优化结果和构效关系, 可以认为化合物9是个里程碑式的化合物, 比较理想的候选药物。但是9在实验动物体内发生广泛代谢, 需要证明代谢产物是否为活性成分所致。为此设计了用3H标记的化合物9和14C标记的胆固醇, 用肠套管和胆道改道的大鼠模型 (多么复杂而精细的整体实验!), 评价代谢产物的活性以及9或活性代谢产物的作用部位, 结果表明3-位苯基上的甲氧基被代谢为羟基 (化合物19) 是一个活性代谢产物, 还证明9和19作用部位是肠壁和肠腔处抑制了促进胆固醇吸收的转运蛋白, 而不是ACAT酶, 这也解释了为什么对ACAT的抑制作用与降低胆固醇没有相关性 (Clader JW. JMed Chem, 2004, 47: 1)。化合物19还可能有其他的氧化代谢产物, 为此合成了一些氧化产物如1位的4-羟基苯基, 3位苯丙基的对位羟基化, 苯环α碳的羟 基化 (R和S构型) 和羰基化, 化合物20~25化合物中, 侧链上S-羟基的化合物23活性最强。
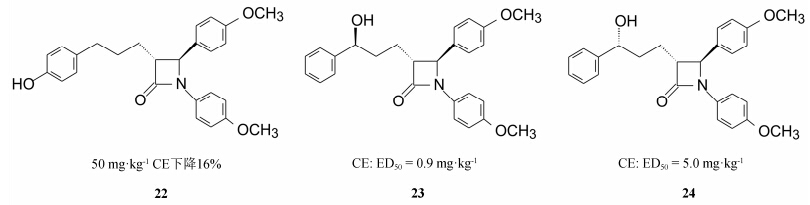
根据上述的构效关系, 再对1位和3位苯基做不同取代, 得到化合物26, 4-位苯环的对位酚羟基和3-位侧链苯环α位S-构型“预制的”羟基不仅是化合物9的代谢产物, 而且也是活性的重要基团。为阻止1-位和3-位苯环的代谢作用, 化合物26结构中还引入可阻止代谢的氟原子, 26的化学结构和构型使药 效和药代性质达到最佳配置, 口服生物利用度F = 35%~65%, 血浆蛋白结合率90%, 半衰期t1/2 = 19~30 h, 肾脏排泄11%, 粪排泄78%。因而作为候选化合物, 命名为依折麦布 (ezetimibe) 进入临床研究。

经过三期临床研究, 依折麦布于2002年被批准上市, 临床用于阻止机体对膳食中胆固醇的吸收, 可单剂应用或与他汀或贝特类药物合用, 治疗高胆固醇症。