 2014, Vol. 49
2014, Vol. 49

早在1938年生理学家就发现动物致敏后体内 产生不稳定的化学物质, 称作慢反应物质,1960年Brockelhurst进一步证明这是免疫组织在受到抗原攻击产生的特殊物质, 可引起平滑肌 (如气道和十二指肠) 的缓慢收缩, 称之为过敏性慢反应物质 (SRS-A) (Brocklehurst WE,et al. J Physiol,1960,151: 416- 435)。随着分离方法 (如HPLC) 和结构鉴定技术的进步,1979年瑞典Samuelsson等证明了SRS-A主要是由3种物质组成: 白三烯C4、D4和E4 (LTC4、LTD4、LTE4), 分别是由谷胱甘肽、胱甘二肽和半胱氨酸经硫醚键相连的20碳四烯酸, 白三烯可引起平滑肌强烈收缩, 增加肺泡的通透性, 增加黏膜上皮细胞的分泌等, 导致哮喘和过敏性疾病。
白三烯作为内源性物质, 是由花生四烯酸经氧化代谢生成含有环氧乙烷结构的LTA4, 经由谷胱甘肽开环生成LTC4, 进而依次降解成LTD4和LTE4, 他们都是致炎因子, 其中LTD4的活性最强。图 1是LTC4、LTD4和LTE4的结构。

|
Figure 1 白三烯的化学结构 |
用同位素标记白三烯 (例如[3H-LTD4]) 的实验表明, 它们结合在肺和支气管的炎症细胞膜上, 与特定的膜受体结合, 该受体称作半胱氨酸白三烯受体1 (CysLT1)。阻断CysLT1功能可抑制气道炎症引起的哮喘和过敏症。
2 初始研究 2.1 以色甘酸为先导物的结构优化1980年以前, 人们研究慢反应物质的拮抗剂是用致敏豚鼠的肺中提取的渗出液引起平滑肌收缩作为筛选模型评价化合物的抑制活性, 发现并证明了色酮化合物FPL-55712 (1) 具有解除上述收缩活性 (Appleton RA,Bantick JR,Chamberlain TR,et al. J Med Chem,1977,20: 371-379; Adams III GK,Lichenstein LM. Nature,1977,270: 255-257)。
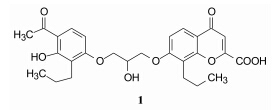
默克公司是研究白三烯的化学及生物功能的先驱之一,1980年开始研究白三烯拮抗剂。该公司合成了LTD4作为工具药, 得以用体外引起豚鼠平滑肌收缩模型评价化合物活性。分析化合物1和LTD4的化学结构, 共有的结构是羧基、共轭双键、疏水基团等, 说明当初在不明配体结构的情况下,1隐含了白三烯的某些结构特征。公司筛选样品库, 发现化合物2有中等强度的拮抗活性。他们将LTD4、化合物1和2作为设计LTD4受体拮抗剂的苗头结构。
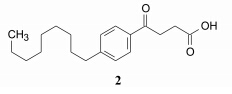
通过融合化合物1和2的结构片段, 合成了以化合物3和4为代表的活性化合物, 分子中与色酮环相连的羧基相当于LTD4中氨基酸的羧基, 烷链上的羧基相当于二十碳四烯酸中的羧基, 化合物3的正壬苯基和化合物4的含共轭三烯的烃基链相当于LTD4的共轭疏水片段。3和4的体外活性强于1和2, 但体内未呈现活性, 而且容易发生代谢作用。

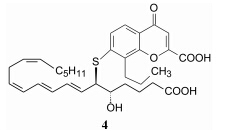
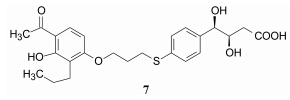
代谢不稳定性主要源于色酮片段, 将其更换成苯丁酸, 保持化合物的左端不变, 合成了一系列化合物, 有代表性的是化合物5, 具有体外抑制受体CysLT1的功能和体内阻止LTD4诱导平滑肌的收缩作用。但5仍容易被代谢失活, 是因为在肝脏中酮基先被还原成羟基, 继而发生β-氧化生成低活性的代谢产物6。

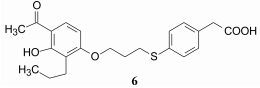
为了提高5的代谢稳定性, 在结构修饰中发现7具有体外和体内活性, 与[3H-LTD4]竞争结合受体的IC50 = 1 μmol·L-1, 灌胃哮喘大鼠的EC50 = 1 mg·kg-1, 而且在消化道是稳定的。7进入临床研究, 发现对肝脏产生不良反应, 终止了研究 (Jones TR,Young RN,Champion E,et al. Can J Physiol Pharmacol,1986,64: 1068-1075)。
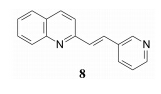
虽然以色甘酸为线索未能研发成药物, 但积累了药物化学和药理学经验, 默克继续前行。由于活性评价的限制, 不能做海量筛选, 公司的药物化学家凭借LTD4拮抗剂的构效关系知识, 对化合物结构用肉眼作直觉性挑选, 之后进行活性评价。在筛选的一万多个样品中发现了喹啉化合物8, 离体活性IC50 = 6 μmol·L-1, 灌胃哮喘大鼠有效剂量3 mg·kg-1。虽然这些数据逊于化合物1, 但作为新的结构类型有修饰的潜力, 例如分子尺寸较小, 可加入含羧基的侧链, 以及形成氢键的基团等。
对化合物8进行结构变换。吡啶环被苯环替换, 活性不变, 但3-羟基苯化合物 (9) 活性显著提高,IC50 = 0.56 μmol·L-1, 而相应的2-羟基或4-羟基化合物没有活性 (IC50 > 50 μmol·L-1)。说明苯环上3-羟基对受体结合的重要性, 此外, 羟基还为引出羧酸侧链提供了连接“把手”。

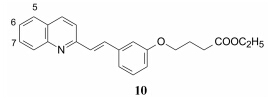
LTD4受体拮抗剂的酸性基团是必需的药效团特征, 为了引入羧基, 经氧原子合成了一系列烷酸和烷酸酯, 体外和大鼠体内活性试验表明, 化合物10的IC50 = 0.58 μmol·L-1, 灌胃大鼠有效剂量为1.5 mg·kg-1, 化合物10作为先导化合物进行优化。
为了避免未取代的喹啉环在体内发生氧化代谢而出现特质性毒性 (ITT), 在环上连接化学致钝取代基。通过对化合物10与LTD4结构的分子叠合, 发现在喹啉环的5、6和7位还存在容纳亲脂性基团的空间, 为此, 分别用亲脂性氯原子取代, 结果表明7-氯取代物 (11) 活性最强,IC50 = 39 nmol·L-1, 灌胃大鼠0.5 mg·kg-1剂量的抑制率为41%。

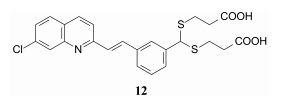
化合物11的进一步优化是引入第二个酸性侧链, 以模拟LTD4结构中含有两个酸性的特征。模拟LTD4的硫醚结构, 以二硫代缩醛引出两个烷酸链, 其中化合物12活性提高,IC50 = 3 nmol·L-1, 大鼠灌胃0.15 mg·kg-1剂量的抑制率为43%。12的大鼠和猴的药代性质良好, 但对鼠猴 (squirrel monkey) 的活性不佳, 究其原因是亲脂性的双酸与血浆白蛋白的结合率高达99.9%, 血液中游离药物水平低, 难以在靶细胞中达到足够的有效浓度。
为了克服11的不足, 提高向细胞内的分布, 优化的策略是将羧基 (一个或两个) 换成酰胺基。结果表明化合物13的活性显著增高,IC50 = 0.8 nmol·L-1, 灌胃大鼠ED50 = 0.07 mg·kg-1。13的代号为MK-571, 进入开发阶段。13对大鼠和猴有良好的口服吸收和生物利用度, 中等水平的血浆结合率, 适宜的半衰期, 以及对多种致敏动物的支气管收缩有解痉作用 (Zamboni R,Belley M,Champion E,et al. J Med Chem, 1992, 35: 3832-3844)。

化合物13完成了临床前试验, 进入临床研究,I和II期临床试验结果令人满意。然而并行实验的小鼠和大鼠长时间和大剂量用药, 发现动物肝脏重量增加, 过氧化物酶体的酶活性增高, 这些征候对于长期用药的患者可能导致肝癌。基于安全性考虑, 中止了MK-571的研发。
3.6 光学活性化合物: 未成功的挽救MK-571含有1个手性碳原子, 当初临床应用的是消旋物, 虽然临床前研究表明,R和S构型的活性和安全性没有显著性差异, 但为了挽救MK-571的不幸, 通过实验深入比较了两个对映异构体的动物安全性, 结果表明, 在高剂量下S异构体引起动物肝脏增重, 血清转氨酶升高, 而R异构体不影响肝脏, 并且R的药动学优于S构型。这样在中止了一年临床试验之后,1990年又开展了R构型的MK-571临床研究, 并命名为韦鲁司特 (verlukast)。然而, 在顺利地进行II期研究中, 发现 有3% 的受试患者转氨酶升高, 从而又不得不停止了研发。
4 备份候选化合物: 孟鲁司特的成功 4.1 单硫醚化合物新药创制需要有备份的候选化合物 (back up drug), 默克公司在研发韦鲁司特的同时, 准备了备份药物。设定的目标是: 化学结构类型不同于韦鲁司特; 有更强的活性; 改善的药动学性质等。
在韦鲁司特结构中, 连接喹啉环和苯环键的双键和二硫代缩醛具有潜在的不稳定性, 在光照下双键与硫原子可发生分子间缩合反应。为消除该隐患, 将双键 -CH=CH- 变换为 -CH2-CH2-、-CH2-O- 和 -CH2-S- 等连接基, 并且去除酰胺侧链上的硫原子, 同时还对烷基链进行烷基取代或嵌入苯环等结构变换, 在合成众多类型的化合物中, 发现14和15的体外活性很高,IC50分别为1.5 nmol·L-1和0.5 nmol·L-1,15强于韦鲁司特 (IC50 = 1 nmol·L-1)。
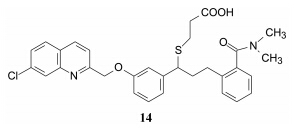

化合物14和15虽然活性强于韦鲁司特, 但在大鼠体内被迅速清除, 半衰期短, 分析原因是酰胺发生了水解作用 (韦鲁司特的酰胺不易水解, 反映出不同的结构代谢样式的差异)。所以, 需要活性和代谢稳定性同时优化, 为此, 同时评价化合物的IC50和大鼠的清除速率。当酰胺片段用 其他极性或非极性基团替换, 仍保持高活性; 含硫醚的羧酸侧链的α-碳被甲基取代可提高活性, 例如化合物16具有代谢稳定性, 并且仍保持对LTD4受体 的高抑制活性。

化合物16含有两个手性碳, 分离出4个单体, 发现硫醚碳为R构型的两个差向异构体可诱发过氧化酶体活性 [以鼠肝的脂肪酸辅酶A氧化酶 (FACO) 为指标], 具有安全性隐患, 而S构型的两个化合物无诱导作用。例如小鼠灌胃800 mg·kg-1的两个R构型的诱导酶活性增高作用为 +128% 和 +155%, 而S构型化合物仅为 -7% 和 +10%, 表明连接硫醚的手性碳为S构型安全性高。然而对LTD4受体的拮抗作用硫醚手性碳的R构型化合物强于S构型, 相差6倍和24倍。这种活性与安全性的矛盾经综合考虑, 确定硫醚手性碳为S构型的化合物为优选结构 (Labelle M,Prasit P,Belley M,et al. Bioorg Med Chem Lett,1992, 2: 1141-1146)。
4.4 候选化合物孟鲁司特的确定对硫醚烷酸链的优化, 是在羧基的α和β碳上分别或同时用甲基或乙基取代 (包括构型的变化), 评价化合物对LTD4受体的拮抗作用、大鼠灌胃诱导FACO活性和肝脏增重作用, 结果表明, 化合物17抑制LTD4活性IC50 = 0.5 nmol·L-1, 不影响FACO的活性, 肝脏增重 +16%。对各种实验动物的药代动力学研究表明, 有良好的口服生物利用度、适宜的半衰期等药动学性质 (Labelle M,Belley M,Gareau Y,et al. Bioorg Med Chem Lett, 1995, 5: 283-288)。遂确定化合物17为韦鲁司特备份候选物, 命名为孟鲁司特 (montelukast), 经Ⅲ期临床试验, 证明用其钠盐10 mg和5 mg咀嚼片可有效地控制哮喘和呼吸道过敏症。1998年FDA批准上市, 成为继扎鲁司特 (zafirlukast,1997上市) FDA批准的第二个LTD4受体拮抗剂。

孟鲁司特作为备份候选药物获得了成功, 而原研药物韦鲁司特因潜在的安全性问题中止于II期临床。