 2014, Vol. 49
2014, Vol. 49



酪氨酸激酶是一类催化ATP上γ-磷酸转移到蛋白酪氨酸残基上的激酶,能够催化多种底物蛋白质酪氨酸残基磷酸化,在细胞生长、增殖、分化中具有非常重要的作用[1, 2]。因此,抑制酪氨酸激酶的活性,可以抑制细胞增殖,是抗肿瘤药物的重要靶点之一。法米替尼是江苏恒瑞医药自主研发的抗肿瘤新药,是一个口服多靶点受体酪氨酸激酶抑制剂,并且具有抗增殖和抑制血管生成的双重抗肿瘤作用[3, 4]。目前,法米替尼作为治疗肾癌的一类新药正处于III临床研究中。
生理药动学 (physiologically based pharmacokinetics,PBPK) 模型是建立在机体的生理、生化、解剖和药物热力学性质基础上的一种整体模型,它将每个相应的组织器官单独作为一个房室看待,房室间借助于血液循环连接[5]。基于生理药动学模型的特点,它可以预测组织器官中药物浓度、描述病理生理参数变化对药物处置的影响以及将在动物中获得的实验结果外推至人,从而预测药物在人体内的药动学以及药物-药物相互作用等,有着非常重要的临床指导意义[6, 7]。药物在人体中的组织分布是不可能通过临床试验获得的,但通过PBPK模型可以预测,从而了解药物在人体中的组织分布以及各组织中的蓄积情况。本文采用GastroPlus软件建立法米替尼的大鼠和猴的PBPK模型,从而预测法米替尼在人体中的药动学及组织分布,为今后进一步药物-药物相互作用研究提供参考。
材料与方法 数据来源大鼠、猴和健康志愿者的血药浓度数据来自文献[3]和本实验室的前期研究,不同种属的体重、给药剂量、给药方式以及血浆蛋白结合率如表 1所示。
|
|
Table 1 The weight,dose,routes of administration,and plasma protein binding (PB) of different species |
采用GastroPlus软件 (Simulations Plus,Inc.,California,United States) 的permeability model根据预测的Caco-2的Papp参数,对法米替尼在不同种属体内的渗透性参数进行了模拟和计算。采用GastroPlus软件的PKPlus模块对法米替尼在大鼠和猴体内的血药浓度采用非房室模型计算药动学参数,并且利用GastroPlus软件的PBPK模块建立法米替尼的大鼠和猴的PBPK模型 (表 2和图 1),首先比较灌流组织限速模型和透膜组织限速模型从而确定最佳的组织限速模型,然后比较实测和模拟的药时曲线,建立并优化大鼠和猴的PBPK模型。
|
|
Table 2 Tissues of blood flow and volume of rat,monkey,and human in the PBPK model. Q: Blood flow; V: Volume |

|
Figure 1 Physiologically based pharmacokinetic model of famitinib in rat,monkey,and human (routes of administration: intravenous,intravenous,and oral,respectively; CLH: Hepatic clearance; CLR: Renal clearance) |
通过计算得到的法米替尼在大鼠和猴体内的血浆清除率,来推算人体内的血浆清除率。不同种属之间的清除率 (CL) 推算,主要采用异速增长公式 (公式1),通常情况下异速增长指数 (b) 的经验值为0.75[8]。但Chiou等[9]研究总结了54种药物的大鼠推算至人的血浆清除率异速增长指数,结果显示平均异速增长指数为0.66。采用GastroPlus软件根据公式2[10]对法米替尼在不同种属的稳态表观分布容积 (Vss) 进行了计算和模拟。通过GastroPlus软件优化好的大鼠和猴的PBPK模型 来预测法米替尼在人体内的药时曲线、药动学参数和组织分布。
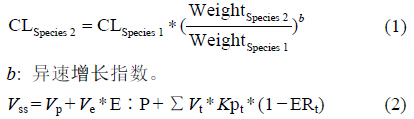
Vp: 血浆容积; Ve: 红细胞容积; E∶P: 红细胞与血浆的浓度比; Vt: 组织容积; Kpt: 组织与血浆的分配系数; ERt: 组织提取率。
结果 1法米替尼的理化性质根据法米替尼的化学结构式 (图 2),采用ACD/ ADME Suite和SimCYP软件测得法米替尼的理化 性质,相对分子质量为410.48,LogP为3.29,pKa (酸) 为14.7,pKa (碱) 为9.2,渗透性 (Caco-2 Papp) 为7.6×10-6 cm·s-1,B/P为1.42。

|
Figure 2 Chemical structure of famitinib |
采用Caco-2的Papp作为数据来源,模拟得到的法米替尼在大鼠、猴和人中的渗透性分别为3.10×10-4、6.87×10-4和6.77×10-4 cm·s-1。根据公式2计算得到法米替尼在大鼠、猴和人的Vss分别是10.2、8.36和9.24 L·kg-1。对大鼠和猴的PBPK模型的组织限速模型考察,结果显示灌流组织限速模型要明显优于透膜组织限速模型。采用非房室模型和优化后的PBPK模型得到的大鼠和猴的静脉给予法米替尼后的药动学参数,大鼠的AUC0-∞实测值和计算值分别为637.9和640.6 µg·h·L-1,实测值与计算值的比值为1.00。猴的AUC0-∞ 实测值和计算值分别为875.4和900.6 µg·h·L-1,实测值与计算值的比值为0.97。结果表明,优化后的大鼠和猴的PBPK模型计算的药动学参数十分接近实测值。从图 3中可以看出大鼠和猴的PBPK模型模拟的药时曲线能够很好地拟合实测的药时曲线。

|
Figure 3 The predicted plasma concentration (solid line) compared to observed data (points) of famitinib in rats (A) and monkeys (B) receiving famitinib intravenous (iv) at 1.88 and 0.75 mg·kg-1,respectively |
利用非房室模型计算的大鼠和猴的法米替尼血浆清除率,采用公式1进行种属外推至人,计算得到的人的法米替尼血浆清除率如表 3所示。根据计算得到的人的法米替尼血浆清除率,在优化后的大鼠和猴的法米替尼PBPK模型的基础上,建立人的PBPK模型,实测和预测的人的药动学参数以及药时曲线如表 3和图 4所示。结果显示,实测AUC0-∞为1 094.9 µg·h·L-1,当进行种属外推 (大鼠→人) 的异速增长指数为0.75和0.66时,预测AUC0-∞ 分别为296.5和671.6 µg·h·L-1,实测值与预测值的比值分别为3.69和1.63。种属外推从猴→人时,预测AUC0-∞为697.7 µg·h·L-1,实测值与预测值的比值为1.57。通过人的PBPK模型,预测法米替尼在人体中的组织 (肺、肝、脾、肾、心) 分布见图 5。
|
|
Table 3 The observed and predicted pharmacokinetic parameters of famitinib in human receiving famitinib oral at 0.42 mg·kg-1 |

|
Figure 4 Predicted plasma concentrations (solid line) compared to observed data (points) for a human oral dose of 0.42 mg·kg-1. A: Rat to human,b = 0.75; B: Rat to human,b = 0.66; C: Monkey to human,b = 0.75 |

|
Figure 5 Predicted tissue distribution of famitinib in human after 0.42 mg·kg-1 oral dose. Cp: Plasma concentration; Ct: Tissue concentration |
人体作为一个整体的系统来处置药物,药物在机体的某部位的动力学的变化会通过血液循环来影响药物在其他部位的变化。近十年来,关于PBPK模型的大量文献报道应用于种属外推、预测药物的体内分布以及药物-药物相互作用等[11, 12]。随着PBPK模型在药物研发领域和监管机构 (如FDA[13]) 推荐的应用更加深入和不断发展,在药物研发过程中将起到越来越重要的作用,尤其是在设计安全有效的临床给药方案以及避免药物-药物相互作用等方面具有非常重要的指导意义。
通常情况下灌流组织限速模型适用于脂溶性高的化合物[14],法米替尼的LogP为3.29,属于脂溶性较高的化合物,模拟结果显示灌流组织限速模型要明显优于透膜组织限速模型。建立并优化法米替尼的大鼠和猴的PBPK模型,对法米替尼的种属外推至人至关重要。通过优化好的大鼠和猴的PBPK模型计算法米替尼在大鼠和猴体内的药动学参数以及药时曲线,从图 3可以看出,优化好的大鼠和猴的PBPK模型可以很好地拟合实测的药动学参数以及药时曲线,结果表明大鼠和猴的PBPK模型建立成功,可以进一步进行种属外推。
通常认为采用PBPK模型进行种属外推以及药物-药物相互作用等研究时,实测值与预测值 (预测值与实测值) 的比值小于2时,表明该模型的计算是被认可和接受的[14, 15]。在优化好的大鼠和猴的PBPK模型的基础上进行种属外推至人时,可以看出当大鼠→人的异速增长指数为0.66时,AUC0-∞的实测值与预测值的比值为1.63 (≤2),并且从图 4可以看出PBPK模型预测的药时曲线能够很好地拟合实测的药时曲线。当猴→人的异速增长指数为0.75时,AUC0-∞的实测值与预测值的比值为1.57 (≤2),并且从图 4可以看出PBPK模型预测的药时曲线能够很 好地拟合实测的药时曲线。异速增长指数通常采用的是经验值,然而对于不同的药物来说,异速增长指数差别较大,文献[9]总结了54种药物的大鼠外推至人的清除率异速增长指数的平均值为0.66,其范围从0.123到1.064,对于不同药物的异速增长指数差别较大,因此0.66只能作为异速增长指数的经验值和探索值,种属外推只是为进一步的研究提供参考。
药物在人体中的组织分布是不可能通过临床试验获得的,但通过PBPK模型可以预测 (图 5)。美国FDA在2012发布的药物相互作用研究指导原则草 案[16]中,推荐利用PBPK软件来预测药物的相互作用,根据其结果来判断,是否需要启动相应的临床试验来验证药物相互作用,因此可以节约大量研发费用和时间。
综上所述,法米替尼的大鼠和猴的PBPK模型建立成功,并且在此基础上进行种属外推至人,能够很好地预测人体内的药动学和组织分布,为今后进一步研究药物-药物相互作用提供了参考。
| [1] | Madhusudan S, Ganesan TS. Tyrosine kinase inhibitors in cancer therapy [J]. Clin Biochem, 2004, 37: 618-635. |
| [2] | Arora A, Scholar E. Role of tyrosine kinase inhibitors in cancer therapy [J]. J Pharmacol Exp Ther, 2005, 315: 971- 979. |
| [3] | Xie C, Zhou JL, Guo ZT, et al. Metabolism and bioactivation of famitinib, a novel inhibitor of receptor tyrosine kinase, in cancer patients [J]. Br J Pharmacol, 2013, 168: 1687-1706. |
| [4] | Zhou AP, Zhang W, Chang CX, et al. Tolerability of famitinib malate: the preliminary results from phase I clinical trial [J]. Chin J New Drugs (中国新药杂志), 2011, 20: 1678-1683. |
| [5] | Reddy MB, Clewell III HJ, Lave T, et al. Physiologically based pharmacokinetic modeling[M]: a tool for understanding ADMET properties and extrapolating to human [M]. NY, USA: InTech, 2013: 197-215. |
| [6] | Jones H, Rowland K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development [J]. CPT Pharmacometrics Syst Pharmacol, 2013, 2: e63–75. doi: 10.1038/psp.2013.41 |
| [7] | Francis CP Law, He SX. The development and application of physiologically based pharmacokinetic modelling [J]. Acta Pharm Sin (药学学报), 1997, 32: 151-160. |
| [8] | Toutain PL, Bousquet A. Plasma clearance [J]. J Vet Pharmacol Ther, 2004, 27: 415-425. |
| [9] | Chiou WL, Robbie G, Chung SM, et al. Correlation of plasma clearance of 54 extensively metabolized drugs between humans and rats: mean allometric coefficient of 0.66 [J]. Pharm Res, 1998, 15: 1474-1479. |
| [10] | Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution [J]. J Pharm Sci, 2002, 91:129-156. |
| [11] | Zhang L, Zhang YD, Zhao P, et al. Predicting drug-drug interactions: an FDA perspective [J]. AAPS J, 2009, 11: 300-306. |
| [12] | Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science [J]. Annu Rev Pharmacol Toxicol, 2011, 51: 45-73. |
| [13] | Zhao P, Rowland M, Huang SM. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions [J]. Clin Pharmacol Ther, 2012, 92: 17-20. |
| [14] | Jones HM, Gardner IB, Watson KJ. Modelling and PBPK simulation in drug discovery [J]. AAPS J, 2009, 11: 155- 166. |
| [15] | Buck SS, Sinha VK, Fenu LA, et al. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs [J]. Drug Metab Dispos, 2007, 35: 1766-1780. |
| [16] | U.S. Food and Drug Administration. Guidance for Drug Interactions Study [S]. 2012. |