 2014, Vol. 49
2014, Vol. 49

2. 第二军医大学药学院 新药研究中心, 上海 200433

 , ZHANG Da-zhi1
, ZHANG Da-zhi1
2. Research Center of New Drug, School of Pharmacy, Second Military Medical University, Shanghai 200433, China
真菌的耐药问题越来越严重[1],已成为临床抗真菌治疗失败的主要原因。抗真菌新药的研发速度远不及真菌产生耐药的速度。抗真菌药物的联合用药虽然作为临床抗耐药真菌的方法,但应用的范围非常有限。文献[2,3,4,5]报道了大量没有抗真菌活性的小分子能显著恢复抗真菌药物对耐药真菌的敏感性,以此寻找抗真菌药物增效剂,是抗耐药真菌研究的新策略。
前期,作者运用耐药性白念珠菌进行抗真菌增效活性筛选,在数个化合物库中,小檗碱 (berberine) 对氟康唑的抗真菌增效作用最为显著: 合用1.0~2.0 μg·mL-1的小檗碱,使氟康唑对40多株临床白念珠菌耐药株的MIC80值都从128.0 μg·mL-1下降为1.0~2.0 μg·mL-1 [6]。进一步的作用机制研究表明,小檗碱能抑制耐药白念株菌对罗丹明-6G的外排量[7],还可导致大量ROS产生,对白念珠菌造成氧化损伤[8],但其作用靶点不明确。研究其作用靶点对耐药真菌的药物研究具有重要意义,出于靶点研究所需的分子探针设计的需要,本文对小檗碱进行基于协同氟康唑抗耐药真菌活性的构效关系进行了研究。
虽然已有大量小檗碱构效关系的报道[9,10],但小檗碱协同氟康唑抗耐药真菌的构效关系研究未见报道。已有的基于小檗碱其他多种生物活性的构效关系研究[11,12],大多以保持小檗碱基本结构不变的结构修饰衍生化为主 (图 1),改变小檗碱基本骨架的研究很少。
 |
Figure 1 The reported structure derivatization of berberine |
本课题借鉴了C13烷基化的修饰方法,设计合成了13-苄基取代的衍生物。由于小檗碱的刚性平面结构,使其溶解性、生物利用度和作为全身用药的成药性均较差,本研究期望通过改变基本骨架,不仅可能克服其上述缺点,还有可能发现新结构,因此,还设计合成了N-苄基取代的异喹啉类化合物,模拟打开小檗碱D环的结构,探索新结构骨架。
通过与取代苄溴化合物反应,在小檗碱13位引入基团,形成13-取代小檗碱1a~1e (合成路线1)。其中化合物1c、1d未见文献报道。
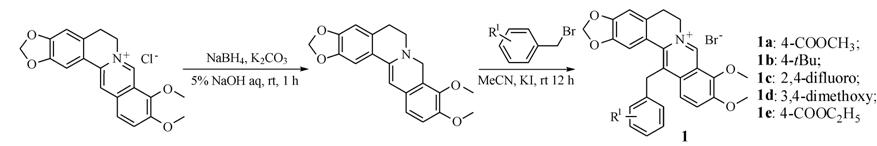
Scheme 1 The synthetic routes of compounds 1a-1e
以胡椒乙胺为起始原料,与羧酸缩合成酰胺,经Bischler-Napieralski反应[13],环合形成二氢异喹啉 类衍生2a~2d (合成路线2); 以胡椒乙胺为起始原 料,分别与甲醛、苯甲醛、4-硝基苯甲醛发生Pictet- Spengler反应[14],环合形成3个四氢异喹啉类衍生物,再分别进行烷基化可以获得两类衍生物 (合成路线3): ① 通过N烷基化形成3a~3f; ② 依次与苄溴和碘甲烷反应,形成四氢异喹啉盐类衍生物4a和4b。其中化合物3b、3c、3d、3e、3f、4a未见文献报道。
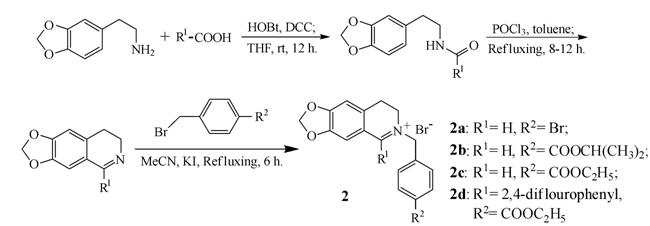
Scheme 2 The synthetic routes of compounds 2a-2d
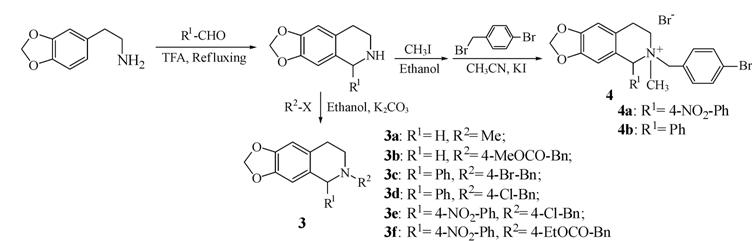
Scheme 3 The synthetic routes of compounds 3a-3f,4a and 4b
结果与讨论 1 化合物的合成目标化合物经1H NMR和MS确证,理化性质及光谱数据见表 1。
|
|
Table 1 Physical and spectra data of the title compounds |
抑菌浓度分数指数 (fractional inhibitory concentration index,FICI) 是评价联合用药的两药相互作用方式的主要参数,为每一种药物联合抑菌时所需最低抑菌浓度 (MIC) 与单用时MIC的比值之和。FICI ≤ 0.5表示二者有协同作用,0.5 < FICI < 4表示二者无关。
从表 2可以看出,化合物1a~1d单用时,MIC80为32.0、64.0 μg·mL-1,表现了微弱的抗真菌活性; 当它们与氟康唑 (8.0 μg·mL-1) 合用时,MIC80降低为2.0、1.0 μg·mL-1,FICI分别为0.13、0.13、0.08、0.09,均表现了与氟康唑非常好的协同作用,与对照药小檗碱相当。说明从小檗碱13位碳引入取代苄基仍能维持其协同氟康唑抗耐药真菌的活性。
|
|
Table 2 Interaction of fluconazole and berberine analogues against clinical isolates of C. albicans 103 resistant to fluconazole by the microbroth dilution assay. FCZ 8.0 μg·mL-1 |
二氢异喹啉类化合物2a~2d的结构中,保持了小檗碱的A、B、C、E等4个环的结构,类似打开 了小檗碱的D环结构。其中,活性较好的两个化合 物2b和2c与氟康唑 (8.0 μg·mL-1) 合用时的MIC80分别为32.0、16.0 μg·mL-1,但活性显著低于小檗碱 (1.0 μg·mL-1)。2a、2d和2e则无活性。四氢异喹啉类化合物3a~3f均无协同活性; 季铵盐4a和4b 的设计,相当于恢复小檗碱结构中的季铵离子,虽然4b具有一定的协同作用,但活性低于小檗碱,且4a没有活性。综合上述对小檗碱的结构改造研究,所合成的异喹啉类衍生物的活性总体低于小檗碱结构修饰类化合物。结果提示,改变小檗碱的基本骨架结构,可以发现具有协同氟康唑抗耐药白念珠菌活性的化合物,虽然活性较低,但值得深入研究。
3小结本课题通过对小檗碱的构效关系初步研究表明,向小檗碱13位碳引入取代苄基后,获得的衍生物均表现了与小檗碱相当的协同氟康唑抗耐药真菌活性,提示可以此方式引入各种功能基团,用于其构效关系和分子探针设计研究; 小檗碱结构改造研究表明,模拟打破小檗碱D环,所设计的N-苄基异喹啉类化合物2b、2c和4b表现了一定的活性,提示虽然小檗碱的基本骨架对维持其活性非常重要,但并非不可改变,值得进一步的结构优化研究。小檗碱的药理作用非常广泛,其构效关系研究大多集中于维持其基本骨架不变的结构修饰研究,本课题的研究结果为小檗碱的其他生物活性的构效关系研究提供一种新的结构改造方法。
实验部分 1 化学合成色谱硅胶板: HSGF254硅胶板,烟台芝罘黄务硅胶开发试验厂; 柱色谱填料: 硅胶H (200~300目),烟台芝罘黄务硅胶开发试验厂; 中性氧化铝 (100~200目): 中国医药集团上海化学试剂责任有限公司; 1H NMR: Bruker DRX-300型; LC-MS: Agilent LC/ MS-6120 (ESI-MS); 原料与溶剂分别购置于阿拉丁试剂 (上海) 公司和中国医药集团上海化学试剂责任有限公司。
1.1 小檗碱衍生物1a~1e的合成将NaBH4 (0.30 g,7.5 mmol) 溶解于5% NaOH溶液(5 mL) 后,滴加含小檗碱 (3.71 g,10 mmol) 与碳酸钾 (3.60 g,30 mmol) 的甲醇 (125 mL)溶液中,室温搅拌1 h; 抽滤收集析出的黄绿色固体,滤饼经30% 乙醇 (20 mL) 与80% 乙醇 (20 mL) 洗涤后,用乙醇重结晶得到黄色粉末二氢小檗碱 (2.50 g,74%)。
称取二氢小檗碱 (674 mg,2 mmol) 与4-溴苄溴 (500 mg,2 mmol) 于100 mL的圆底烧瓶内,用乙腈 50 mL溶解,加入催化量的KI后于室温搅拌12 h; 待反应完全后过滤,滤液减压浓缩除去溶剂,残渣用适量无水乙醚洗涤3次; 残渣烘干后用二氯甲烷/甲醇体系 (100∶1) 进行中性氧化铝柱色谱分离,最终得黄色粉末溴代-13-(4-溴代苯甲基)-小檗碱1b (714 mg,61%)。1a和1c~1e的合成均用以上合成方法合成。
1.2 二氢异喹啉衍生物2a~2d的合成称取胡椒乙胺 (16.52 g,100 mmol)、HOBt (13.50 g,100 mmol) 和DCC (20.60 g,100 mmol) 于1 L的圆底烧瓶内,加入THF (100 mL) 使其溶解; 称取甲酸 (4.60 g,100 mmol) 用THF (100 mL) 溶解后缓慢滴加于上述圆底烧瓶内,待滴加完毕后于室温搅拌12 h,TLC监
视反应完全程度,待反应完全后过滤除去白色不溶物;滤液经减压浓缩除去溶剂,残渣用二氯甲烷 (100 mL) 溶解,饱和Na2CO3溶液洗涤3次,无水MgSO4干燥,待干燥后过滤,滤液经二氯甲烷/甲醇体系 (80∶1) 硅胶柱色谱纯化,最终得白色固体粉末N-甲酰-(3,4-亚甲二氧基) 苯乙胺 (10.33 g,53%)。
称取N-甲酰-(3,4-亚甲二氧基) 苯乙胺 (1.93 g,10 mmol) 于100 mL的圆底烧瓶内,加入甲苯 (50 mL) 使其充分溶解,在冰浴下缓慢滴加POCl3 (7.67 g,50 mmol); 待POCl3滴加完毕后于回流状态下反应8~12 h,待反应完全后,冷却至室温; 将上述反应液倒入100 mL冰水溶液,用浓氨水调节pH至8~11,用二氯甲烷 (50 mL) 提取上述水溶液3次,合并后用无水MgSO4干燥; 待上述溶液干燥后过滤,滤液经二氯甲烷/甲醇体系 (80∶1) 硅胶柱色谱纯化,最终得微黄色固体粉末6,7-亚甲二氧基-3,4-二氢异喹啉 (835 mg,48%)。
称取6,7-亚甲二氧基-3,4-二氢异喹啉 (350 mg,2 mmol) 与4-溴苄溴 (500 mg,2 mmol) 于100 mL 的圆底烧瓶内,加入乙腈 (50 mL) 后于回流状态下反应6 h; TLC跟踪反应进程,待反应完全后减压浓缩除去溶剂,残渣经二氯甲烷/甲醇 (80∶1) 硅胶柱色谱纯化,最终得黄色固体粉末溴代-2-(4-溴苯甲基)- 6,7-亚甲二氧基-3,4-二氢异喹啉2a (289 mg,34%)。2b~2d的合成均用以上合成方法合成。
1.3 四氢异喹啉3a~3f及四氢异喹啉盐类衍生物 4a,4b的合成称取胡椒乙胺 (16.52 g,100 mmol) 与37% 的甲醛水溶液 (8.12 g,100 mmol) 于250 mL的圆底烧瓶内,在冰浴下加入三氟乙酸 (50 mL); 将反应液于回流状态下反应12 h,TLC监视反应完全程度; 待反应完全后将反应液倒入100 mL冰水中,用 5 mol·L-1的NaOH水溶液调节pH值于8~13,二氯甲烷提取3次,合并提取液后用无水MgSO4干燥,过 滤,滤液经二氯甲烷/甲醇体系 (80∶1) 硅胶柱色谱纯化,最终得白色固体6,7-亚甲二氧基-1,2,3,4-四氢异喹啉 (13.01 g,73%)。以苯甲醛和4-硝基苯甲醛替代甲醛,运用上述方法分别获得1-苯基-6,7-亚甲二氧基-1,2,3,4-四氢异喹啉和1-(4-硝基苯基)-6,7-亚甲二氧基-1,2,3,4-四氢异喹啉用于下一步研究。
称取6,7-亚甲二氧基-1,2,3,4-四氢异喹啉 (1.77 g,10 mmol) 于50 mL的圆底烧瓶内,加入K2CO3 (1.38 g,10 mmol) 与溶解有CH3I (1.42 g,10 mmol) 的乙醇 (50 mL); 反应液于回流状态下反应8 h,TLC监视反应完全程度,待反应完全后过滤除去白色不溶物,滤液减压浓缩; 残渣经二氯甲烷/甲醇 (80∶1) 硅胶柱色谱纯化,最终得白色固体2-甲基-6,7-亚甲二氧基-1,2,3,4-四氢异喹啉3a (1.37 g,72%)。其他四氢异喹啉衍生物的合成均用以上合成方法合成。
按照合成3a的甲基化方法,分别制备1-(4-硝基苯基)-2-甲基-6,7-亚甲二氧基-1,2,3,4-四氢异喹啉和1-苯基-2-甲基-6,7-亚甲二氧基-1,2,3,4-四氢异喹啉。称取1-(4-硝基苯基)-2-甲基-6,7-亚甲二氧基-1,2,3,4-四氢异喹啉 (312 mg,1 mmol) 于100 mL的圆底烧瓶内,加入4-溴苄溴 (500 mg,2 mmol)、乙腈 (50 mL) 与催化量的KI,反应液与于回流状态反应8 h,TLC监视反应完全程度; 待反应完全后,减压浓缩除去溶剂,残渣经二氯甲烷/甲醇 (80∶1) 硅胶柱色谱纯化,最终得白色固体溴代1-(4-硝基苯基)-2-甲基-2-(4-溴苯甲基)-6,7-亚甲二氧基-1,2,3,4-四氢异喹啉4a (428 mg,89%)。用上述方法,以1-苯基-2-甲基-6,7-亚甲二氧基-1,2,3,4-四氢异喹啉合成4b。
2生物活性实验氟康唑 (fluconazole,FCZ) 注射液由大连辉瑞药业有限公司提供; 盐酸小檗碱 (berberine chloride,BBR) 由上海长海医院提供; 二甲基亚砜 (dimethyl sulphoxide,DMSO) 中国医药集团上海化学试剂公司出品; Multiskan MK3型酶标检测仪 (芬兰Labsystems产品); MJX型智能霉菌培养箱 (宁波江南仪器厂); SW-CT-IF型超净化工作台 (苏州安泰空气技术有限公司); 临床分离耐药白色念珠球菌103由长海医院菌种保存中心赠送。
取无菌96孔板,于每排1号孔加RPMI 1640液体培养基100 µL作空白对照; 3~12号孔各加新鲜 配制的菌液100 µL,菌液浓度为每毫升1×103~5×103 CFU。2号孔分别加菌液160 µL和受试化合物溶液40 µL; 12号孔不含药物,只加菌液100 µL作阳性生长对照。2~11号孔进行倍比稀释,使各孔的受试化合物终质量浓度分别为64.0、32.0、16.0、8.0、4.0、2.0、1.0、0.5、0.25和0.125 μg·mL-1,各孔中DMSO含量均低于1%,氟康唑的终质量浓度为8.0 μg·mL-1。
| [1] | Goffeau A. Drug resistance: the fight against fungi [J]. Nature, 2008, 452: 541-542. |
| [2] | Sharma M, Manoharlal R, Negi AS. Synergistic anticandidal activity of pure polphenolcurcumin I in combination with azoles and polyenes generates reactive oxygen species leading to apoptosis [J]. FEMS Yeast Res, 2010, 10: 570-578. |
| [3] | Khan MSA, Malik A, Ahmad I. Anti-candidal activity of essential oils alone and in combination with amphotericin B or fluconazole against multi-drug resistant isolates of Candida albicans [J]. Med Mycol, 2011, 50: 33-42. |
| [4] | Faria NCG, Kim JH, Goncalves LAP. Enhanced activity of antifungal drugs using natural phenolics against yeast strains of Candida and Cryptococcus [J]. Lett Appl Microbiol, 2011, 52: 506-513. |
| [5] | Fujita K, Fujita T, Kubo I. Anethole, a potential antimicrobial synergist, converts a fungistatic dodecanol to a fungicidal agent [J]. Phytother Res, 2007, 21: 47-51. |
| [6] | Quan H, Cao YY, Xu Z, et al. Potent in vitro synergism of fluconazole and berberine chloride against clinical isolates of Candida albicans resistant to fluconazole [J]. Antimicrob Agents Chemother, 2006, 50: 1096-1099. |
| [7] | Zhu SL, Yan L, Zhang YX, et al. Berberine inhibits fluphenazine- induced up-regulation of CDR1 in Candida albicans [J]. Biol Pharm Bull, 2014, 37: 268-273. |
| [8] | Xu Y, Wang Y, Yan L, et al. Proteomic analysis reveals a synergistic mechanism of fluzole and berberine against fluconazole-resistant Candida albicans: endogenous ROS augmentation [J]. J Proteome Res, 2009, 8: 5296-5304. |
| [9] | Li B, Zhu WL, Chen KX. Advances in the study of berberine and its derivatives [J]. Acta Pharm Sin(药学学报), 2008, 43: 773-787 |
| [10] | Liu Y, Xiao C, Wang Y, et al. Synthesis, structure-activity relationship and in vitro anti-mycobacterial evaluation of 13-n- octylberberine derivatives [J]. Eur J Med Chem, 2012, 52: 151-158. |
| [11] | Huang ZJ, Zeng Y, Lan P, et al. Advances in structural modifications and biological activities of berberine: an active compound in traditional Chinese medicine [J]. Mini-Rev Med Chem, 2011, 11: 1122-1129. |
| [12] | Bi CW, Zhang CX, Li YB, et al. Synthesis and structure- activity relationship of cycloberberine as anti-cancer agent [J]. Acta Pharm Sin(药学学报), 2013, 48: 1800-1806 |
| [13] | Park KS, Kang KC, Kim JH, et al. Differential inhibitory effects of protoberberines on sterol and chitin biosyntheses in Candida albicans [J]. J Antimicrob Chemother, 1999, 43: 667-674. |
| [14] | Tóth J, Dancsó A, Blaskó G, et al. 1, 7-Electrocyclization reactions of stabilized α, β: γ, δ-unsaturated azomethineylides [J]. Tetrahedron, 2006, 62: 5725-5735. |