 2019, Vol. 39
2019, Vol. 39 

NMR技术开始用于定量分析始于1963年[1-2],近几年以来,核磁共振定量技术(qNMR技术)在药物含量测定领域的应用越来越广泛[3-6]。1H qNMR的基础是化学环境不同的氢原子核的共振峰面积只与所含的氢原子数目有关。qNMR技术的最大优点是它只需用已知纯度的化合物作为参比物,而无需特定的对照品,只要被测物和参比物中至少分别有1个或1组特征的且互不重叠的共振峰存在,则无需引入任何校正因子,可直接根据共振峰的面积求出所代表的氢原子的数目[6]。
二乙酰吗啡,是以吗啡生物碱作为合成起点得到的半合成毒品,最初其被用做戒除吗啡毒瘾的药物,后来发现它具有比吗啡更强的药物依赖性,即成瘾性更强。长期吸食、注射海洛因可导致人脑区功能异常[7],产生决策行为能力缺失与认知障碍[8],且难以戒断[9-10]。海洛因依赖已成为危害个人健康、家庭和谐与社会稳定的世界性难题[11]。二乙酰吗啡对照品在毒品检测试剂的质控、禁毒工作及药物依赖性研究工作中有重要作用,本文针对二乙酰吗啡建立了1H qNMR定量方法,用于在对照品标定过程中进行辅助定值。
1 仪器与试剂Bruker AVANCE Ⅲ 600核磁共振仪(Bruker公司);METTLER TOLEDO XPE-26电子天平(METTLER TOLEDO公司,d=0.001 mg)。
氘代氯仿(纯度99.8%,含0.03%四甲基硅烷,Cambridge Isotope Laboratories,Inc.);氘代二甲基亚砜(DMSO-d6,纯度99.9%,含0.03%四甲基硅烷,Cambridge Isotope Laboratories,Inc.);对苯二甲酸二甲酯(DMT,纯度100.0%,中国食品药品检定研究院);马来酸(纯度99.94%,Sigma-Aldrich);二乙酰吗啡(纯度98.3%,中国食品药品检定研究院)。
2 方法与结果 2.1 供试溶液的制备精密称取内标物DMT约2 mg及二乙酰吗啡约15 mg至离心管中,再加入氘代氯仿1 mL,震荡摇匀后,转入核磁管中待测。
2.2 检测条件采用noesyigld1d脉冲序列在恒温(25 ℃)下获取1H NMR谱。射频中心(O1P)δ 6.5,谱宽(SWH)15 ppm,驰豫延迟时间(D1)25 s,采样时间(AQ)3.64 s,增益(RG)32,采样点数(TD)64 k,采样次数(NS)32次,空扫次数(DS)4次。
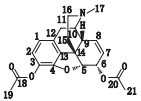
2.3 测定结果采集图谱采集到的1H NMR数据用topspin 3.5.7软件处理,测定样品对内标DMT选定峰峰面积的相对比值,按内标法[12]52计算含量。二乙酰吗啡样品及内标的氢谱如图 1所示,氢谱解析见表 1。

|
1、3.内标的氢信号(peaks of internal standard)2.样品的定量氢信号(quantitation peak of sample) 图 1 二乙酰吗啡及内标的氢谱图 Figure 1 1H NMR spectrum of diacetylmorphine and internal standard |
|
|
表 1 二乙酰吗啡的氢谱解析 Table 1 1H NMR analysis of diacetylmorphine |
将1份样品图谱反复积分5次考察手动积分的精密度,以δ 6.76的单氢信号峰与内标对苯二甲酸二甲酯的定量峰δ 8.10面积的比值计算RSD为0.07%。将1份样品反复测定5次,以δ 6.76的单氢信号峰与内标对苯二甲酸二甲酯的定量峰δ 8.10面积的比值计算RSD为0.22%。显示该方法具有量好的精密度。
2.5 重复性试验平行制备供试溶液5份,按“2.2”项下条件进行测定,以内标法进行计算,二乙酰吗啡的含量按
|
|
表 2 含量测定结果 Table 2 Determination results by qNMR method |
据文献[6]报道,1H NMR用于精确定量时,信噪比(S/N)需满足250,对“2.5”项下最小称样量为14.317 mg的样品的信噪比进行计算,结果为14 985,满足精确定量的要求,计算定量下限为0.239 mg。
2.7 定量结果配制5份平行供试溶液,按照“2.2”项下条件进行测定,按“2.5”项下公式计算二乙酰吗啡的含量,测定结果见表 2,含量平均值为98.7%。
2.8 与其他方法的比较利用质量平衡法[13]计算二乙酰吗啡的含量,公式为含量=(100%-干燥失重-炽灼残渣量)×纯度。二乙酰吗啡样品的干燥失重结果为0.21%,炽灼残渣结果为0.07%,高效液相色谱纯度为98.97%,气相色谱纯度为98.58%,采用液相色谱纯度按质量平衡法计算二乙酰基吗啡的含量为(100%-0.21%-0.07%)×98.97%=98.7%,采用气相色谱纯度按质量平衡法计算二乙酰基吗啡的含量为(100%-0.21%-0.07%)×98.58%=98.3%。采用DSC法[12]83测定的结果为98.4%。采用核磁定量结果为98.7%。计算不同方法定值结果的RSD为0.21%,由此可见3种定量方法得出的4个结果接近,可相互佐证。
3 讨论 3.1 内标及溶剂的选择内标物应具有较高的纯度,较少的质子数,主要吸收峰与样品峰之间无干扰,且不与样品发生反应。最初拟采用DMSO作溶剂,马来酸作为内标,从实验结果可知,二乙酰吗啡发生了降解,主要生成O6-单乙酰吗啡,这是因为二乙酰吗啡3位的酯键在酸性条件下不稳定,易水解脱除乙酰键所致。从二乙酰吗啡和DMT的核磁共振氢谱图中可以看出,DMT苯环上的氢(δ 8.10)和二乙酰吗啡1位上的氢(δ 6.76)不重叠,且内标物的峰稳定,不与样品反应,满足定量要求。因此,本实验选择DMT作内标。样品二乙酰吗啡与内标DMT均易溶于氘代氯仿,溶剂峰不干扰测定;且相对于DMSO-d6,氘代氯仿没有吸湿性,因此能保持干燥,更能避免二乙酰吗啡的水解,利于样品的稳定,故采用氘代氯仿为溶剂。
3.2 内标物与被测物定量峰的选择采用NMR技术对物质进行定量时,待测物和内标物通常会有多个峰。用于定量的内标峰和待测峰应选择与其他峰完全分离,附近基线平坦的峰,没有杂质峰的干扰。因此本文选取了DMT苯环上的峰δ 8.10作为内标定量峰,以二乙酰吗啡的峰δ 6.76为被测组分的定量峰。
3.3 D1的选择通过翻转恢复试验对所有的质子共振谱线的弛豫时间T1值分别进行测试,结果如图 2所示,内标峰δ 8.10的T1为3.765 s,样品定量峰δ6.76的T1为2.860 s。据文献报道,在进行qNMR试验时,为了保证积分结果的准确性,驰豫延迟时间D1不应小于T1的5倍[14],为保证结果的准确性,最终D1值选定为25 s,最终结果表明,该选择可以满足测量精度的要求。

|
A.内标峰δ 8.10的T1(T1 of internal standard at δ 8.10)B.样品峰δ 6.76的T1(T1 of sample at δ 6.76) 图 2 内标及二乙酰吗啡的T1 Figure 2 T1 of IS and diacetylmorphine |
在标准物质的标化过程中,含量确定是非常关键的步骤。特别是进行首批对照品的标定时,往往需要用不同原理的方法进行赋值,以保证结果的准确性。
质量平衡法(mass balance method)又叫杂质扣除法[15-16],即从100%中减去与主成分结构类似杂质、水分、残留溶剂和不挥发性杂质的量。质量平衡法通常被认为具有很高准确度,并且被世界卫生组织(WHO)[17-18]、欧洲药典[13]推荐为药品标准物质定值方法。质量平衡法含量测定过程较为复杂,理论上需要明确样品中的各种杂质,在日常工作中其最常用HPLC法进行纯度测定,常遇到的情况是无法完全确定样品中的所有杂质,这时就可能会遇见杂质的响应因子与主成分有差异,杂质在检测条件下不出峰或者待测样品的色谱峰与有关物质的色谱峰之间分离度较差等情况,从而引入一定的误差。
DSC纯度测定是基于共熔体系熔点降低原理,主成分和杂质如能形成共熔体系,则主成分的熔点会随着杂质的摩尔百分比含量的提高而逐渐降低。此方法样品用量少,操作简便,不需要对照品,不需分离杂质,能测定物质的绝对纯度。但是其也有很多限制,如它不适用于非共熔体系,不适用于熔融分解的物质,也不适合纯度过低的样品。
1H qNMR法的原理是化学环境不同的氢原子核的共振峰面积只与所含的氢原子数目有关。因为大部分有机分子都含有氢元素,因此它有较高的普适性;它样品制备简单,由于氢原子的信号很强,检测十分快速;无需特定的对照品,信号强度仅与氢原子数目相关,无需引入校正因子,特异性强,同时还可以提供化合物的结构信息,可以同时完成样品的定量和定性分析。当然,1H qNMR法也具有其局限性,当化合物结构复杂,不具备1个或1组特征的且与其他峰基线能完全分离的共振峰时,则不适合采用此种方法进行精确定量,但可以采用其他原子核的qNMR法进行检测。qNMR的另一个巨大的优势是在一次实验中既能给出定量信息,同时也能给出十分可靠的定性信息,可以在实验条件的优化上获得更明确的方向。比如本文在最初拟采用DMSO作溶剂,马来酸作为内标,采集数据后即可从图谱中观察到二乙酰吗啡发生了降解,通过核磁信号解析可知主要生成了O6-单乙酰吗啡,进而可知是二乙酰吗啡3位的酯键水解脱除乙酰键所致,说明它极易水解,酸性的内标以及吸湿性的溶剂都可能有不利的影响。利用这些信息,可以很快地知道如何有针对性地优化条件。
质量平衡法、DSC法、1H qNMR法原理不同,可相互补充,相互佐证。本文建立了对二乙酰吗啡含量的核磁共振分析方法,此方法快速简便可靠。同时还采用质量平衡法及DSC含量测定方法测定其含量,测定结果一致。
| [1] |
JUNGNICKEL JL, FORBES JW. Quantitative measurement of hydrogen types by integrated nuclear magnetic resonance intensities[J]. Anal Chem, 1963, 35(8): 938. DOI:10.1021/ac60201a005 |
| [2] |
HOLLIS DP. Quantitative analysis of aspirin, phenacetin, and caffeine mixture by nuclear magnetic resonance spectrometry[J]. Anal Chem, 1963, 35(11): 1682. DOI:10.1021/ac60204a043 |
| [3] |
马晓丽, 邹萍萍, 雷伟, 等. 定量核磁技术参数的优化及其在中草药定量分析领域的应用[J]. 药学学报, 2014, 49(9): 1248. MA XL, ZOU PP, LEI W, et al. Optimization of experimental parameters for quantitative NMR(qNMR)and its application in quantitative analysis of traditional Chinese medicines[J]. Acta Pharm Sin, 2014, 49(9): 1248. |
| [4] |
LIU X, KOLPAK MX. Automatic analysis of quantitative NMR data of pharmaceutical compound libraries[J]. Anal Chem, 2012, 84(15): 6914. DOI:10.1021/ac301544u |
| [5] |
LINDON JC, NICHOLSON JK. Analytical technologies for metabonomics and metabolomics, and multi-omic information recovery[J]. Trends Anal Chem, 2008, 27(3): 194. DOI:10.1016/j.trac.2007.08.009 |
| [6] |
BHARTI SK, ROY R. Quantitative 1H NMR spectroscopy[J]. Trends Anal Chem, 2012, 35: 5. DOI:10.1016/j.trac.2012.02.007 |
| [7] |
QIU YW, HAN LJ, LV XF, et al. Regional homogeneity changes in heroin-dependent individuals:resting-state functional MR imaging study[J]. Radiology, 2011, 261(2): 551. DOI:10.1148/radiol.11102466 |
| [8] |
YUAN K, QIN W, DONG M, et al. Gray matter deficits and resting-stateabnormalities in abstinent heroin-dependent individuals[J]. Neurosci Lett, 2010, 482(2): 101. DOI:10.1016/j.neulet.2010.07.005 |
| [9] |
李强, 王亚蓉, 李玮, 等. 强制戒断的海洛因成瘾者默认功能网络fMRI研究[J]. 实用放射学杂志, 2012, 28(11): 1665. LI Q, WANG YR, LI W, et al. fMRI study of default mode network in heroin-dependent patients during compulsory abstinence[J]. J Pract Radiol, 2012, 28(11): 1665. DOI:10.3969/j.issn.1002-1671.2012.11.001 |
| [10] |
伊涛, 傅先明, 钱若兵, 等. 海洛因成瘾者扣带前回结构与功能连接的MRI研究[J]. 中国微侵袭神经外科杂志, 2011, 16(12): 541. YI T, FU XM, QIAN RB, et al. MRI study of the structure and functional connectivity of anterior cingulate cortex in heroin addicts[J]. Chin J Minim Invasive Neurosurg, 2011, 16(12): 541. |
| [11] |
FISCHER B, OVIEDO-JOEKES E, BLANKEN P, et al. Heroin-assisted treatment(HAT)a decade later:a brief update on science and politics[J]. J Urban Health, 2007, 84(4): 552. DOI:10.1007/s11524-007-9198-y |
| [12] |
中华人民共和国药典2015年版.四部[S]. 2015: 52, 83 ChP 2015. Vol Ⅳ[S]. 2015: 52, 83 |
| [13] |
EP 9.0[S]. 2015: 733
|
| [14] |
HOLZGRABE U, WAWER I, DIEHL B. NMR Spectroscopy in Pharmaceutical Analysis[M]. Oxford: Elsevier Science, 2008: 44.
|
| [15] |
马康, 苏福海, 王海峰, 等. 有机纯度标准物质定值技术研究进展[J]. 分析测试学报, 2013, 32(7): 901. MA K, SU FH, WANG HF, et al. Progress on certified technique for chemical purity of organic reference material[J]. J Instrum Anal, 2013, 32(7): 901. DOI:10.3969/j.issn.1004-4957.2013.07.023 |
| [16] |
MATHKAR S, KUMAR S, BUSTOL A, et al. The use of differential scanning calorimetry for the purity verification of pharmaceutical reference standards[J]. J Pharm Biomed Anal, 2009, 49(3): 627. DOI:10.1016/j.jpba.2008.12.030 |
| [17] |
WHO Technical Reports Series No. 885(Part A), General Guidelines for the Establishment, Maintenance and Distribution of Chemical Reference Substance[S]. 1999
|
| [18] |
World Health Organization. Supplementary Part A(3.3), Evaluation of Chemical Reference Substance, International Pharmacopoeia. Vol 5[S]. 2003
|