

2. 中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室/农业部动物流感重点实验室, 哈尔滨 150069

 , WANG Weiguo1, TANG Xiaoming1, LU Xinghua1, LIN Yuan1, DENG Guohua2, WANG Changjian1
, WANG Weiguo1, TANG Xiaoming1, LU Xinghua1, LIN Yuan1, DENG Guohua2, WANG Changjian1
2. Animal Influenza Key Laboratory of the Ministry of Agriculture, State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Harbin 150069, China
在过去的一个世纪,人类社会共发生过四次流感大流行,分别是1918年的H1N1西班牙流感,1957年的人H2N2流感和1968年的人H3N2流感以及2009年的甲型流感,这几次事件中,禽流感病毒通过感染人等宿主发生了不同程度的抗原漂移[1-4]。H3亚型禽流感病毒能够感染多个物种,导致其容易发生抗原漂移和基因重组,报道称H3亚型禽流感病毒与H5N1、H9N2禽流感病毒通过基因重组产生的H3亚型重组病毒能感染鸭并导致发病[5-6],韩国的一项研究指出犬在感染禽源H3N2流感病毒后出现了严重的呼吸系统疾病[7],英国还报道了H3N8流感病毒感染海豹导致海豹群体出现162例死亡的案例[8]。
学者通过研究发现,中国部分地区散养鸭分离的新型H3N2禽流感病毒的内部基因与H7N3、H7N7以及H7N9亚型禽流感病毒的内部基因表现出非常高的同源性[9-11];鸭源H3N8亚型禽流感病毒具有感染鸡并在鸡群间传播的潜在风险[11]。H3亚型与其他亚型禽流感病毒重组频繁,可能产生能够大规模流行的新型病毒,是危害公共卫生安全的隐患。环洞庭湖地区位于东亚-澳大利亚全球候鸟迁徙路线上,每年有超过百种种类的野鸟来越冬[12],野鸟是禽流感病毒的天然宿主[13],也是禽流感病毒传播和进化的关键环节。本研究对与野鸟接触最为密切的散养鸭进行流调监测,同时对流感病毒重组频繁的活禽市场区域进行采样,对分离的H3亚型禽流感病毒进行全基因测序和进化分析,给我国禽流感综合防控提供数据支持和理论依据。
1 材料与方法 1.1 主要试剂病毒RNA提取试剂盒购自北京天根生化科技有限公司;反转录试剂盒M-MLV购自Promega公司;DNA聚合酶和DL2000 DNA Marker购自TaKaRa公司;胶回收试剂盒购自OMEGA公司;DNA测序试剂盒购自Applied Biosystem公司。
1.2 毒株31株H3亚型禽流感病毒来源于2011—2015年湖南省洞庭湖地区活禽市场与散养禽场的监测采样样品,由中国农业科学院哈尔滨兽医研究所国家禽流感参考实验室分离并保存。
1.3 鸡胚与红细胞9~11日龄SPF鸡胚与鸡血购自哈尔滨兽医研究所实验动物中心,1%鸡红细胞按实验室标准配制。
1.4 全基因测序按照病毒RNA提取试剂盒说明书提取病毒RNA,按照反转录试剂盒说明书将病毒RNA反转录成cDNA。参照Hoffmann等[14]的引物,应用RT-PCR反应扩增病毒的8个基因片段。用胶回收试剂盒回收PCR产物中的目的片段,按测序试剂盒说明书进行病毒的全基因组序列测定。
1.5 遗传进化分析测序结果用DNASTAR软件中的Seqman程序进行拼接,NCBI数据库在线BLAST比对,选择参考序列,经MegAlign比对后,利用MEGA5.0分析特殊位点的氨基酸,绘制各基因片段进化树。
1.6 基因型划分挑选不同分离时间、分离地点、HA与NA基因具有差异性的病毒进行全基因的比较,应用MEGA5.0中Bootstrap method将各基因序列比对1 000次,根据病毒的同源性差异和在进化树中的不同分支,将核苷酸相似性高于95%的病毒分到同一组,根据PB2、PB1、PA、HA、NP、NA、M和NS八个基因片段的相关性给病毒划分基因型。
2 结果 2.1 H3亚型AIV的病毒分离情况31株H3亚型禽流感病毒于2011—2015年分离自湖南省活禽市场与散养鸭场的监测样品,包括18株H3N2,13株H3N8;31株分离株中8株病毒为散养禽场拭子分离,23株为活禽市场拭子样品分离(毒株名称与详细分离情况见表 1)。
|
|
表 1 洞庭湖地区H3亚型禽流感病毒分离情况 Table 1 Separation of H3 subtype avian influenza virus in Dongting Lake area |
基因序列比对结果显示,各病毒HA基因全长均为1 701 bp,编码567个氨基酸。去除16个氨基酸的信号肽之后,31株H3的HA基因的裂解位点为325EKQ/HTR↓GLFG333,为低致病性禽流感病毒分子特征;受体结合位点为98Y、138A、183H、190E、194L、224R、226Q、227S、228G,不存在使病毒毒力增强以及使病毒倾向结合人源受体的氨基酸位点;所有病毒均存在5个潜在的糖基化位点,分别为22NGT24、38NAT40、165NVT167、285NGS287、483NGT485,仅A/duck/Hunan/S42712/2014(H3N8)一株病毒含有2NLS4糖基化位点。对各病毒的NA基因与参考序列进行比对,有两株H3N2病毒A/duck/Hunan/S11727/2015(H3N2)与A/environment/Hunan/S10958/2015(H3N2)的N2基因在第62—64位缺失3个氨基酸。
2.3 H3亚型AIV的内部基因分子特征根据31株病毒的宿主差异与分离时间、地点的差异性以及HA与NA基因的差异性选出21株病毒进行全基因的测序。氨基酸比对结果显示,21株H3亚型禽流感病毒的PB2基因无E627K、D701N、T271A等使病毒毒力增强或使病毒获得感染哺乳动物能力的氨基酸位点突变;M2基因不存在V27I、L26F、S31N等能使病毒抗金刚烷胺与金刚乙胺的氨基酸位点突变;各病毒的NS1基因无缺失现象;各病毒的PB1-F2基因全长均编码90个氨基酸,无N66S等使病毒增强对哺乳动物致病性的位点突变,其他基因片段也无特殊氨基酸位点的突变。
2.4 H3亚型AIV的各基因片段的进化分析 2.4.1 HA基因的进化分析根据进化树中的位置将各病毒分为北美分支与欧亚分支,本研究中的所有HA基因均位于欧亚分支上,核苷酸的相似性在87.1%~100.0%,氨基酸的相似性在93.3%~ 100.0%,各病毒进一步可以分为9个不同的Group,具有明显的遗传多样性,Group 1和9中病毒与越南的野鸟源H3N8病毒相似性较高,Group 5和6与泰国和蒙古地区的野鸟源H3N6病毒相似性较高,可能有相同的进化来源;其他分组的病毒则与国内浙江或江西等地的家禽H3病毒相似性较高(图 1A)。

|
A. HA; B. NA(N2);C. NA(N8);N.北美分支;E.欧亚分支;▲.散养鸭场分离毒 A. HA; B. NAC N2); C. NACN8); N. North American lineage; E. Eurasian lineage; 图 1 H3亚型禽流感病毒的HA与NA基因的进化树 Figure 1 Phylogenetic analyses of HA and NA genes of H3 avian influenza viruses |
本研究中的18株H3N2病毒的N2基因根据进化树中位置与相似性可分成5个分组,均位于欧亚分支上,核苷酸相似性在80.6%~100.0%之间,氨基酸的相似性在82.8%~100.0%,多数病毒位于Group 4与5,与浙江省等湖南省周边省份的H3N2病毒的同源性较高,其他病毒位于Group 1~3,分别与H10N2、H9N2、H5N2等有较高的相似性(图 1B)。13株H3N8的核苷酸相似性为76.7%~100.0%,氨基酸相似性为85.6%~100.0%,仅A/duck/Hunan/S11933/2013(H3N8)与A/duck/Hunan/S1256/2012(H3N8)位于欧亚分支上,与H3N8、H11N8病毒有较高的相似性;其他病毒位于北美分支,不同组的病毒与众多亚型的N8病毒位于相同分支,与越南、韩国的野鸟源N8病毒也有较高的相似性,来源相对复杂(图 1C)。
2.4.3 内部基因的进化分析PB2基因可分为10个不同的分组(图 2),各病毒间的核苷酸相似性为87.1%~99.9%,氨基酸的相似性为98.2%~100.0%,其中A/chicken/Hunan/S11685/2015(H3N2)与越南2007年从野鸟分离的H5N1病毒具有较高的相似性,一株2011年从环境样品中分离的A/environment/Hunan/S4350/2011(H3N8)与国家禽流感病毒参考实验室在2013年分离的H5N1病毒相似性非常高,表明该病毒可能为H5N1病毒基因重组的内部基因供体,处于其他分组的病毒则与湖南省周边省份的低致病性禽流感亚型病毒在来源上更加相近,主要以H4、H6、H11毒株为主;PB1、PA基因与PB2基因的进化树结构相似,同时发现部分从散养家禽分离得到的病毒与越南、韩国、蒙古等周边国家的野鸟分离的H3、H11毒株处于相同分组;NP基因可分为7个不同的分组,各病毒间的核苷酸相似性为87.7%~99.7%,氨基酸的相似性为96.0%~100.0%,Group 2中几株病毒与越南的H12N5毒株、韩国的H4N6毒株、日本的H6N8毒株均有较高的相似性,Group 1中病毒与越南H3N8毒株有较高的相似性,其他分组的病毒与国内家禽分离的多种低致病性禽流感亚型相近;各病毒M基因的核苷酸相似性为93.5%~99.9%,氨基酸相似性为96.6%~100.0%,分为6个不同分组,Group 3中病毒与越南鸭源H7N1处于相同分组,其他组则与国内家禽低致病性亚型相似性较高;NS基因分为Allele A与Allele B分支,仅A/duck/Hunan/S1256/2012(H3N8)处于Allele A分支,在进化来源上与A/Goose/Guangdong/1/1996(H5N1)同处Allele A分支,该病毒与其他位于Allele B分支的病毒的核苷酸相似性低于71.5%,与2009年越南分离的A/duck/Vietnam/G18/2009(H12N5)的相似性为98.8%,可能有相同的进化来源(内部基因进化树见图 2),其他病毒的NS基因位于Allele B分支,在进化来源上与1997年香港人流感病毒同属于Allele B分支病毒。

|
A. PB2; B. PB1; C. NP; D. PA; E. M; F. NS 图 2 H3亚型禽流感病毒的6个内部基因的进化分析 Figure 2 genetic analyses of the six internal genes of H3 avian influenza viruses |
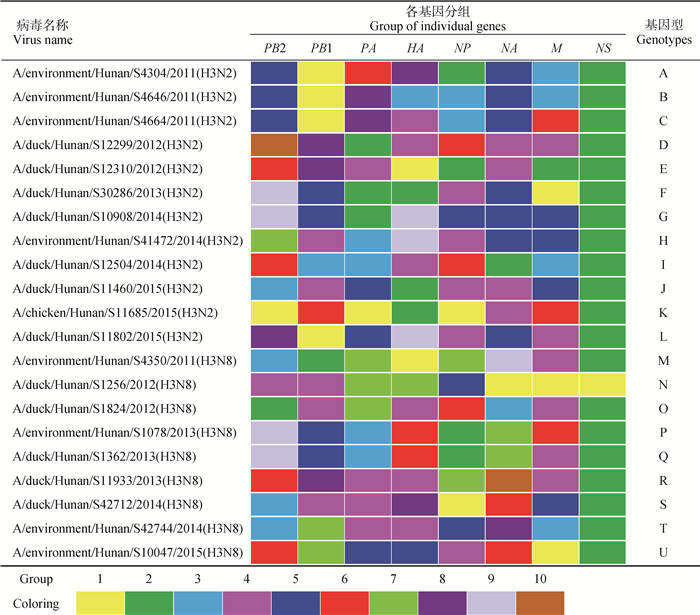
根据病毒的8个基因片段的同源性以及在各进化树中的位置,可以将12株H3N2和9株H3N8共21株病毒分为21个不同的基因型,每株病毒的基因型均不相同。环洞庭湖地区H3亚型禽流感病毒来源复杂,各病毒的不同基因片段间发生了频繁的基因重组,具有明显的遗传多样性(全基因测序毒株名称和基因型见表 2)。
|
|
表 2 H3亚型禽流感病毒毒株名称及基因型划分 Table 2 Viruses and genotypes of H3 avian influenza viruses |
洞庭湖地区位于全球东亚-澳大利亚候鸟迁徙路线上,每年有超过百种的鸟类途经进行避暑和越冬,野鸟与家禽的接触不仅给低致病性禽流感病毒的基因交换创造了有利条件,还可能造成新型流感病毒的产生。本试验中的H3亚型禽流感病毒各基因片段出现了非常复杂的基因重组现象,部分病毒的HA基因不仅与相邻省份家禽分离的H3毒株处于相同分支,且与韩国、越南等周边国家在野鸟中分离的H3毒株同源性非常高,表明洞庭湖地区的H3亚型禽流感病毒处于持续输出和流入的状态。
A/chicken/Hunan/S11685/2015/(H3N2)与越南2007年从野鸟分离的H5N1病毒的PB2、M基因具有较高的同源性,A/environment/Hunan/S4350/2011(H3N8)则与湖南省2013年分离的H5N1病毒的PB2、PA基因同源性非常高,近年在韩国、北美、欧洲地区流行的高致病性禽流感病毒H5N8的基因与H5N1及多种低致病性禽流感病毒存在基因重组情况[15-16],同类研究中也指出H3亚型禽流感病毒的内部基因与H5N1、H7N9、H9N2等病毒均存在不同程度的基因重组现象[17-18],表明H3亚型病毒作为基因供体给新型流感病毒的产生提供了条件。H3亚型病毒能够感染畜、禽、马、海豹以及多种哺乳动物,在传播和进化过程中具有非常大的变异性,本试验研究了2011—2015年洞庭湖地区家禽场的H3亚型禽流感病毒,每过一段时间病毒的基因型均发生了改变,尽管如此,这些改变可能在未来造成的影响仍不清楚,需要进行长期的监测,及时掌握该地区各种亚型禽流感病毒的流行进化情况。
4 结论环洞庭湖地区H3亚型禽流感病毒具有明显的遗传多样性,部分病毒的内部基因与H5和H7亚型禽流感病毒可能发生了基因重组,提示洞庭湖地区的H3亚型禽流感病毒给高致病性禽流感的产生提供了基因供体;分离于散养家禽的部分H3病毒与韩国和日本等周边国家家禽间暴发的H3病毒相似性较高,表明洞庭湖地区的禽流感病毒处于不断输出和流入状态,给新型流感病毒的产生创造了条件,因此,对该地区家禽进行禽流感病毒的持续监测,对于我国禽流感病毒的综合防控具有非常重要的意义。
| [1] | TAUBENBERGER J K, REID A H, KRAFFT A E, et al. Initial genetic characterization of the 1918"Spanish" influenza virus[J]. Science, 1997, 275(5307): 1793–1796. DOI: 10.1126/science.275.5307.1793 |
| [2] | NEUMANN G, NODA T, KAWAOKA Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus[J]. Nature, 2009, 459(7249): 931–939. DOI: 10.1038/nature08157 |
| [3] | SCHOLTISSEK C, ROHDE W, VON HOYNINGEN V, et al. On the origin of the human influenza virus subtypes H2N2 and H3N2[J]. Virology, 1978, 87(1): 13–20. |
| [4] | PAPPAS C, VISWANATHAN K, CHANDRASEKARAN A, et al. Receptor specificity and transmission of H2N2 subtype viruses isolated from the pandemic of 1957[J]. PLoS One, 2010, 5(6): e11158. DOI: 10.1371/journal.pone.0011158 |
| [5] | SONG M S, OH T K, MOON H J, et al. Ecology of H3 avian influenza viruses in Korea and assessment of their pathogenic potentials[J]. J Gen Virol, 2008, 89: 949–957. DOI: 10.1099/vir.0.83462-0 |
| [6] | ZHOU H B, ZHANG A D, CHEN H C, et al. Emergence of novel reassortant H3N2 influenza viruses among ducks in China[J]. Arch Virol, 2011, 156(6): 1045–1048. DOI: 10.1007/s00705-011-0940-0 |
| [7] | SONG D, KANG B, LEE C, et al. Transmission of avian influenza virus (H3N2) to dogs[J]. Emerg Infect Dis, 2008, 14(5): 741–746. DOI: 10.3201/eid1405.071471 |
| [8] | ANTHONY S J, ST LEGER J A, PUGLIARES K, et al. Emergence of fatal avian influenza in New England harbor seals[J]. mBio, 2012, 3(4): e00166–12. |
| [9] | LI C, YU M, LIU L T, et al. Characterization of a novel H3N2 influenza virus isolated from domestic ducks in China[J]. Virus Genes, 2016, 52(4): 568–572. DOI: 10.1007/s11262-016-1323-0 |
| [10] | YANG D Q, LIU J, JU H B, et al. Genetic analysis of H3N2 avian influenza viruses isolated from live poultry markets and poultry slaughterhouses in Shanghai, China in 2013[J]. Virus Genes, 2015, 51(1): 25–32. |
| [11] |
崔鹏飞, 彭志, 张芳, 等. 四株鸭源H3N8亚型禽流感病毒对SPF鸡的感染性分析[J]. 畜牧兽医学报, 2016, 47(6): 1209–1214.
CUI P F, PENG Z, ZHANG F, et al. Infectivity of four H3N8 avian influenza viruses of duck origin in SPF chickens[J]. Acta Veterinaria et Zootechnica Sinica, 2016, 47(6): 1209–1214. (in Chinese) |
| [12] |
钟福生, 王焰新, 邓学建, 等. 洞庭湖湿地珍稀濒危鸟类群落组成及多样性[J]. 生态环境, 2007, 16(5): 1485–1491.
ZHONG F S, WANG Y X, DENG X J, et al. Species diversity of rare, endangered and national key protected waterfowls in Dongting Lake wetlands[J]. Ecology and Environment, 2007, 16(5): 1485–1491. DOI: 10.3969/j.issn.1674-5906.2007.05.032 (in Chinese) |
| [13] | MUNSTER V J, BAAS C, LEXMOND P, et al. Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds[J]. PLoS Pathog, 2007, 3(5): e61. DOI: 10.1371/journal.ppat.0030061 |
| [14] | HOFFMANN E, STECH J, GUAN Y, et al. Universal primer set for the full-length amplification of all influenza A viruses[J]. Arch Virol, 2001, 146(12): 2275–2289. DOI: 10.1007/s007050170002 |
| [15] | KANG H M, LEE E K, SONG B M, et al. Novel reassortant influenza A(H5N8) viruses among inoculated domestic and wild ducks, South Korea, 2014[J]. Emerg Infect Dis, 2015, 21(2): 298–304. DOI: 10.3201/eid2102.141268 |
| [16] | POHLMANN A, STARICK E, GRUND C, et al. Swarm incursions of reassortants of highly pathogenic avian influenza virus strains H5N8 and H5N5, clade 2. 3. 4. 4b, Germany, winter 2016/17[J]. Sci Rep, 2018, 8(1): 15. |
| [17] |
彭志. 2014~2015年环洞庭湖与鄱阳湖散养鸭群禽流感病毒感染情况调查[D].长沙: 湖南农业大学, 2016.
PENG Z. Investigation of infection of Avian Influenza in domestic duck in Dongting Lake and Poyang Lake from 2014 to 2015[D]. Changsha: Hunan Agricultural University, 2016. (in Chinese) http://cdmd.cnki.com.cn/Article/CDMD-10537-1017033537.htm |
| [18] |
关立峥.中国H3N2(2009-2014年)和H5(2015-2016年)亚型禽流感病毒的生物学特性研究[D].兰州: 甘肃农业大学, 2017.
GUAN L Z. Biological characterization of H3N2(from 2009 to 2014) and H5(from 2015 to 2016) avian influenza viruses in China[D]. Lanzhou: Gansu Agricultural University, 2017. (in Chinese) http://cdmd.cnki.com.cn/Article/CDMD-10733-1017824803.htm |