 2017, Vol. 47
2017, Vol. 47
2. 浙江新东港药业股份有限公司, 浙江 台州 318000
2. Zhejiang Neo-Dankon Pharmaceutical Co., Ltd., Taizhou 318000, China
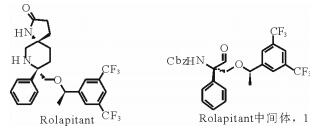
肿瘤已成为严重威胁人类生命及生活质量的一类疾病, 现全球为此而接受放化疗患者超过1 000万人。较同类药物阿瑞匹坦相比[5], 该药半衰期长达180小时(>7天), 口服后吸收迅速, 具有较高的应用价值和广阔的市场前景[6]。其中, 70%~80%患者需要使用P物质/神经激肽-1(NK-1)受体拮抗剂来止吐[1]。Rolapitant是一种新型的NK-1受体拮抗剂, 具有强效、选择性、竞争性的特点[2-6]。
|
(S)-1-((R)-1-(3, 5-二(三氟甲基)苯基)乙氧基)-3-氧-2-苯基丙烷-2-基)氨基甲酸苄酯(1)是合成罗拉匹坦的关键中间体。目前, 文献报道[7]罗拉匹坦中间体的制备方法主要为手性源的合成路线:一般以商业化的N-Cbz-L-苯甘氨酸为原料, 与苯甲醛二甲缩醛缩合得到中间体4, 4与溴甲基化产物6发生取代反应得化合物7, 最后经还原和水解等5步反应得到目标产物1。手性源的工艺路线解决了手性碳光学纯度的问题, 为产业化较理想的选择。但该路线存在以下几个问题:1)4的合成使用了低沸点溶剂乙醚, 安全系数低, 且反应周期长(反应时间为3d), 另外后处理需多次过滤, 操作繁琐, 从而提高了产品的成本[8]; 2)6的合成需溴化氢气体, 现场制备后处理麻烦, 环境污染严重, 溴化氢钢瓶成本较高, 危险性大。因而需对罗拉匹坦的关键中间体1进行研究与优化, 为后续罗拉匹坦的路线优化和避专利路线的开发打下坚实的基础。
1 实验部分 1.1 仪器与试剂仪器:熔点用WRS-1B熔点测定仪测定, 温度未校正。核磁共振氢谱采用Varian-400 MHz核磁共振仪测定, 氘代氯仿作溶剂, 四甲基硅烷作内标; 质谱在Frace DSQ FINNGAN上测定; 产品含量分析采用岛津GC-2010上测定; 比旋光度采用美国鲁道夫旋光仪Autopol Ⅴ测定。
试剂:所有试剂均为市售工业品。
1.2 实验方法 1.2.1 (2R, 4S)-2, 4-二苯基噁唑烷酮-3-甲酸苄酯4的制备在装有机械搅拌和温度计的1 L四口烧瓶中, 氮气氛围下, 加入N-Cbz-L-苯甘氨酸(2, 114 g)、三氟化硼乙醚684 g, 降至0℃, 滴加苯甲醛二甲缩醛(3, 91 g, 0.6 mol), 在0℃~5℃条件下继续反应8 h, 大量浅黄色固体析出, 抽滤, 石油醚打浆洗涤两次, 烘干, 得(2R, 4S)-2, 4-二苯基噁唑烷酮-3-甲酸苄酯4为浅黄色固体137 g, 收率为92%。熔点为:192℃~193℃(lit[8] 194℃~195℃); [a]D25=+162.5°(0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.79 (d, J=12.0 Hz, 1H), 4.96 (d, J=12.0 Hz, 1H), 5.40-5.54 (m, 1H), 6.72-6.83 (m, 3H), 7.13-7.22 (m, 3H), 7.39-7.44 (m, 10H); MS (ESI):m/z (%) 374.3 [M+H]+。
1.2.2 (S)-1-(3, 5-二(三氟甲基)苯基)乙基溴甲基醚6的制备在带有机械搅拌、冷凝管和温度计的0.5 L三口烧瓶中, 依次加入(S)-1-(3, 5-二(三氟甲基)苯基)乙醇(5, 206 g, 0.8 mol)、三聚甲醛(26 g, 0.88 mol), 升温至80℃搅拌20 min, 降至室温, 通入溴化氢气体(氮气鼓入溴化氢醋酸溶液后, 经氯化钙干燥管逸出), GC中控至原料5的含量小于5%(体积分数)后停止反应, 加入正己烷(300 g), 分液除去水相, 无水硫酸钠干燥, 减压抽滤, 浓缩回收溶剂后得棕色油状物粗品, 经油泵减压蒸馏收集73℃的馏分为(S)-1-(3, 5-二(三氟甲基)苯基)乙基溴甲基醚6, 是无色的油状物216 g, 收率77%, 纯度99%。1H NMR (400 MHz, CDCl3) δ: 1.55 (d, J=6.4 Hz, 3H), 4.98 (q, J=6.4 Hz, 1H), 5.43 (d, J=4.8 Hz, 1H), 5.78 (d, J=4.8 Hz, 1H), 7.80 (s, 2H), 7.86 (s, 1H); MS (EI):m/z (%) 351 (16) [M]+, 257 (23), 241 (100)。
1.2.3 内酯化合物7的制备在带有机械搅拌、恒压滴液漏斗和温度计的5 L四口烧瓶中, 依次加入4(130 g, 0.35 mol)、四氢呋喃(3 kg), 氮气氛围下, 冷却至-70℃, 加入1mol/L的KHMDS(350 mL), 保温搅拌1 h后, 缓慢滴加6(123 g, 0.35 mol), 滴毕继续搅拌1 h, 加饱和氯化铵淬灭反应(1 kg), 再用乙酸乙酯萃取(1.5 kg×2), 合并有机相用饱和食盐水洗涤(2 kg×2), 无水硫酸钠干燥, 抽滤浓缩回收溶剂后, 经柱层析(流动相为φ(正己烷):φ(乙酸乙酯)=20:1)得7, 为无色油状物162 g, 收率72%。[a]D25=+42.5° (0.4, MeOH); 1H NMR (400 MHz, CDCl3) δ: 1.57 (d, J=6.4 Hz, 3H), 4.14 (d, J=4.8 Hz, 1H), 4.30 (d, J=4.8 Hz, 1H), 4.79 (d, J=12.0 Hz, 1H), 4.96 (d, J=12.0 Hz, 1H), 4.99 (q, J=6.4 Hz, 1H), 6.72-6.83 (m, 3H), 7.14-7.22 (m, 3H), 7.39-7.43 (m, 10H), 7.79 (s, 2H), 7.88 (s, 1H); MS (ESI):m/z (%) 644.6 [M+H]+。
1.2.4 还原产物T8的制备在装有机械搅拌和温度计的1L四口烧瓶中, 依次加入内酯化合物7(84 g, 0.13 mol)、四氢呋喃(600 g), 氮气氛围下, 冷却至0℃左右, 分批加入四氢铝锂(1.5 g, 0.04 mol), 加毕后, 混合物继续搅拌30 min, 用饱和食盐水淬灭(200 g), 过滤, 分液, 有机相浓缩后得74 g无色液体8, 收率88%, 产品无需纯化直接用于之后反应。
1.2.5 (S)-1-((R)-1-(3, 5-二(三氟甲基)苯基)乙氧基)-3-氧-2-苯基丙烷-2-基)氨基甲酸苄酯T1的制备在带有磁力搅拌、冷凝管和温度计的0.5 L四口烧瓶中, 依次加入还原产物8(65 g), NMP 150 g, 碳酸氢钠(2.5 g)、水2 g, 升至50℃搅拌反应12h, 加入水(500 g), 分液, 水相用乙酸乙酯萃取(300 g×2), 合并有机相用饱和食盐水洗涤(300 g), 无水硫酸钠干燥, 抽滤浓缩回收溶剂, 得黄色油状物粗品, 常规减压蒸馏后得46 g浅黄色油状物1, 收率为85%。[a]D25=+48.9o (0.8, MeOH); 1H NMR (400 MHz, DMSO-d6) δ: 1.34 (d, J=6.4 Hz, 3H), 3.90 (d, J=9.6 Hz, 1H), 4.71-4.72 (m, 1H), 5.09 (s, 2H), 7.33-7.41(m, 10H) 7.90 (s, 2H), 7.99 (s, 1H), 8.35 (s, 1H), 9.53 (s, 1H); MS (ESI): m/z (%)=540.5 [M+H]+。其合成路线如图 1所示。

|
图 1 (S)-1-((R)-1-(3, 5-二(三氟甲基)苯基)乙氧基)-3-氧-2-苯基丙烷-2-基)氨基甲酸苄酯的合成路线 Fig. 1 Scheme of ((S)-1-((R)-1-(3, 5-bis(trifluoromethyl)phenyl)ethoxy)-3-oxo-2-phenylpropan-2-yl)carbamate synthesis |
向带有磁力搅拌的100 mL三颈烧瓶中, 依次加入(S)-1-(3, 5-二(三氟甲基)苯基)乙醇(5, 2.58 g)、三聚甲醛(0.36 g)、二氯甲烷(10 mL)、40%溴化氢水溶液(10 mL), 室温下反应6 h, 加水洗涤(50 mL), 分液, 有机相用无水硫酸钠干燥, 过滤, 旋蒸溶剂后得黄色固体粗品, 石油醚重结晶得二((S)-1-(3, 5-二(三氟甲基)苯基)乙氧基)甲烷9为白色晶体3.96 g, 收率75%, mp:46~47℃; [a]D25=-15.4o (0.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.50 (d, J=6.4 Hz, 6H), 4.66 (s, 2H), 4.89 (q, J=6.4 Hz, 2H), 7.68-7.79 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 23.8 (2C), 73.5 (2C), 91.3, 119.1, 121.5 (2C), 121.8, 124.5, 126.1 (4C), 127.3, 131.8 (q, JC-F=33.1 Hz, 4C), 146.1 (2C); MS (EI):m/z(%) 528 (23) [M]+, 287 (31), 241 (100)。
1.2.7 (S)-1-(3, 5-二(三氟甲基)苯基)乙酸乙酯10的制备向带有磁力搅拌的100 mL三颈烧瓶中, 依次加入(S)-1-(3, 5-二(三氟甲基)苯基)乙醇(5, 2.58 g, 0.01 mol)、三聚甲醛(0.36 g, 0.012 mol)、二氯甲烷(10 mL)和溴化氢醋酸溶液(10 mL), 室温下反应6 h, 加水洗涤(50 mL), 取有机相用无水硫酸钠干燥, 过滤, 旋蒸溶剂后得浅黄色固体粗品, 石油醚重结晶得(S)-1-(3, 5-二(三氟甲基)苯基)乙酸乙酯10为无色晶体2.18 g, 收率80%, m.p.:43~44℃; [a]D25=-23.1° (0.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 1.58 (d, J=6.8 Hz, 3H), 2.12 (s, 3H), 5.95 (q, J=6.4 Hz, 1H), 7.80-7.81 (m, 3H); 13C NMR (100 MHz, CDCl3) δ: 21.1, 22.2, 70.9, 121.8 (2C), 124.5, 126.3 (2C), 131.9 (q, JC-F=33.1 Hz, 2C), 144.4, 169.9; MS (EI):m/z(%) 300 (13) [M]+, 257 (23), 241 (100)。
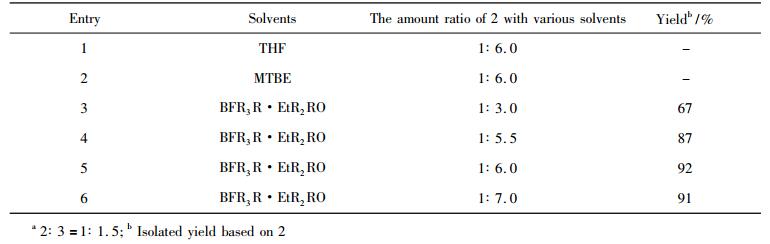
2 实验结果和讨论 2.1 (2R, 4S)-2, 4-二苯基噁唑烷酮-3-甲酸苄酯4的合成条件优化合成4时使用溶剂乙醚[7-8], 由于乙醚沸点低, 极性较小, 其溶解性差, 反应周期长, 笔者开始尝试醚类的代替品。如四氢呋喃(THF)或甲基叔丁基醚(MTBE)等作为反应溶剂, 但其极性及溶解性均大于乙醚, 产物在体系中析不出固体; 最后通过增加三氟化硼乙醚的用量, 当用量为底物2质量的6倍时, 得到较为理想的效果, 收率高达92%, 如表 1所示。由于改善了体系的溶解性, 明显减少溶剂用量, 缩短反应时间, 简化实验操作, 提高反应的安全性。
|
|
表 1 溶剂对4收率的影响a Tab. 1 Effect of solvents on the yield of 4 |
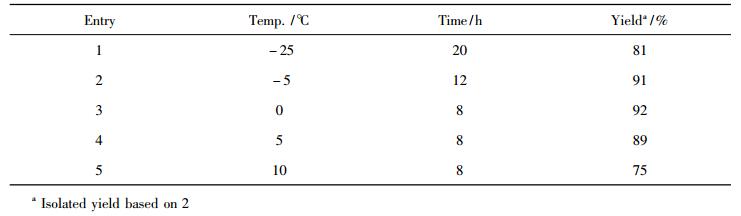
同时, 还考察了反应温度与时间对4收率的影响(表 2), 结果表明, 该步缩合反应较适宜的温度为-5~5℃, 温度太高杂质较多, 温度太低原料反应不完全, 均不利于反应的进行, 最后选取反应温度为0℃, 反应时间为8h, 得到的收率较为理想。
|
|
表 2 反应温度和时间对4收率的影响 Tab. 2 Effect of reation temperature and time on the yield of 4 |
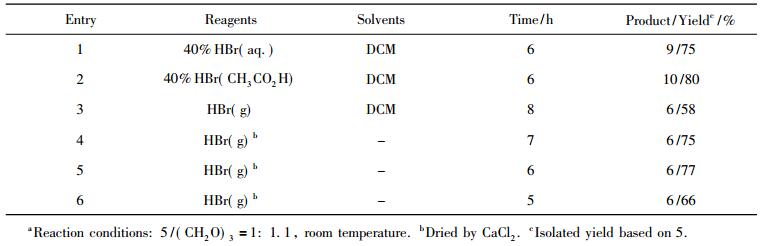
溴甲基化反应类似于氯甲基化反应[9], 不同的是反应试剂溴化氢气体较易氧化成溴单质, 导致反应收率和纯度降低。经过多次试验, 发现影响(S)-1-(3, 5-二(三氟甲基)苯基)乙基溴甲基醚6的合成收率的因素主要是反应试剂、反应溶剂及反应时间。溴甲基化一般需要现场制备溴化氢气体, 目前文献中的方法主要缺点为溴化氢气体腐蚀性大, 对设备要求较高, 且后处理较麻烦, 会产生大量“三废”污染。所以笔者考察了廉价商业化的溴化氢溶液对产物收率的影响, 结果见表 3。
|
|
表 3 不同反应条件对6收率的影响 Tab. 3 Effect of different reaction conditions on the yield of 6a |
从表 3结果看出, 以DCM为溶剂, 40%(质量分数)溴化氢水溶液为溴化试剂, 室温下反应6 h后, 经NMR, MS和旋光度等谱图的确证, 产物为二((S)-1-(3, 5-二(三氟甲基)苯基)乙氧基)甲烷9, 收率为75%。其可能的机理为:首先解离后的甲醛与体系中的氢质子结合形成盐11, 其碳原子的亲电性能增加, 与亲核试剂醇5发生加成, 再失去氢质子成了不稳定的半缩醛12, 接着与氢质子结合形成新的盐13, 失水变成14, 再与一分子醇5加成并失去氢质子, 最后形成缩醛产物9(图 2)。以DCM为溶剂, 40%溴化氢醋酸溶液为溴化试剂, 室温下反应6 h, 经相关谱图的确证, 原料5直接与醋酸发生乙酯化反应, 得到无色的晶体状固体为(S)-1-(3, 5-二(三氟甲基)苯基)乙酸乙酯10, 收率为80%。因而, 笔者在综合参考文献[10-11]的基础上, 尝试以DCM为溶剂, 原料(S)-1-(3, 5-二(三氟甲基)苯基)乙醇和三聚甲醛搅拌溶解后, 氮气鼓入溴化氢醋酸溶液, 逸出溴化氢气体导入反应体系中, 成功制备目标产物6, 但收率中等(58%); 接着, 尝试在无溶剂条件下, 5和三聚甲醛先升温熔融, 再导入干燥的溴化氢气体, 反应收率较为理想(77%), 另外还考察了反应时间对收率的影响, 结果表明, 在无溶剂条件下, 室温下反应6 h, 产物收率高达77%。

|
图 2 二((S)-1-(3, 5-二(三氟甲基)苯基)乙氧基)甲烷的反应机理 Fig. 2 Proposed mechanism of systhesis of bis((S)-1-(3, 5-bis(trifluoromethyl)phenyl)ethoxy)methane |
按照文献[7]的方法, (2R, 4S)-2, 4-二苯基噁唑烷酮-3-甲酸苄酯4在低温条件下与(S)-1-(3, 5-二(三氟甲基)苯基)乙基溴甲基醚6发生取代反应后, 再经四氢铝锂还原后, 顺利得到还原产物8, 最后经水解制得目标产物1。实验考察了碱、反应溶剂、反应温度及时间对水解反应收率的影响(表 4)。
|
|
表 4 不同反应条件对1收率的影响 Tab. 4 Effect of different reaction conditions on the yield of 1 |
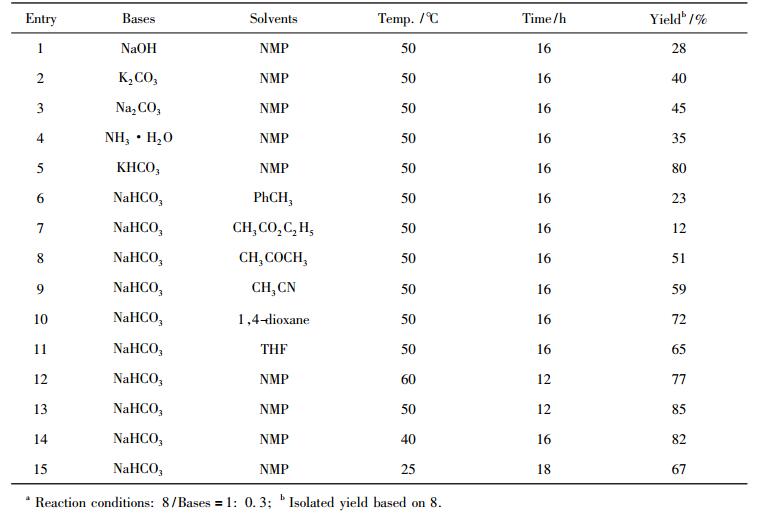
可以看出, 开始选用氢氧化钠、碳酸钠或碳酸钾等中强碱T, 可能碱性较强对反应体系有破坏, T得到的收率较低(28%~45%)。当选用氨水时, 由于稳定性较差, 在50℃反应较长时间, 氨气容易逸出, 导致收率偏低(35%)。当选用碳酸氢钾或碳酸氢钠等无机弱碱时, 收率明显提高。发现亲水性溶剂如N-甲基吡咯烷酮(NMP)作为反应溶剂, 得到较高收率(75%), 选用疏水性溶剂如甲苯或乙酸乙酯等溶剂时, 由于对无机碱碳酸氢钠的溶解性较差, 得到的收率较低。同时, 反应温度对收率也有影响。在室温下反应时, 反应缓慢收率较低, 升至60℃后反应、副反应增多, 收率降低; 因而选择50℃, 反应收率较为理想。由表 4可知, 水解反应的最佳条件为:以N-甲基吡咯烷酮为溶剂, 还原产物8与碳酸氢钠的物质的量比为1:0.3时, 反应温度为50℃, 反应时间为12 h, 收率达85%。
3 结论以N-Cbz-L-苯甘氨酸与苯甲醛二甲缩醛为原料经缩合, 再与溴甲基化的产物发生取代反应, 最后经还原与水解反应制得罗拉匹坦关键中间体, 总收率为50%。实验过程中考察了溶剂与碱的种类及用量、反应温度及催化剂用量等因素对反应的影响, 最终确定了最佳的反应条件, 与之前的工艺相比, 该法具有操作简便、反应条件温和、后处理安全和环境友好等优点。
| [1] |
GRUNBERG S M, HESKETH P J. Control of chemotherapy-induced emesis[J]. N Engl J Med, 1993, 329: 1790-1796. DOI:10.1056/NEJM199312093292408 |
| [2] |
SYED Y Y. Rolapitant: first global approval[J]. Drugs, 2015, 75(16): 1941-1945. DOI:10.1007/s40265-015-0485-8 |
| [3] |
RAPOPORT B L, CHASEN M R, GRIDELLI C, et al. Safety and efficacy of rolapitant for prevention of chemotherapyinduced nausea and vomiting after administration of cisplatinbased highly emetogenic chemotherapy in patients with cancer: two randomized, active-controlled, double-blind phase 3 trials[J]. Lancet Oncol, 2015, 16(9): 1079-1089. DOI:10.1016/S1470-2045(15)00035-2 |
| [4] |
SCHWARTZBERG L S, MODIANO M R, RAPOPORT B L, et al. Safety and efficacy of rolapitant for prevention of chemotherapy-induced nausea and vomiting after administration of moderately emetogenic chemotherapy or anthracycline and cyclophosphamide regimens in patients with cancer: a randomized, active-controlled, double-blind phase 3 trial[J]. Lancet Oncol, 2015, 16(9): 1071-1078. DOI:10.1016/S1470-2045(15)00034-0 |
| [5] |
DIEMUNSCH P, GAN T J, PHILIP B K, et al. Aprepitant-PONV protocol 091 international study group. Singledose aprepitant vs ondansetron for the prevention of postoperative nausea and vomiting: a randomized, double-blind phase Ⅲ trial in patients undergoing open abdominal surgery[J]. Br J Anaesth, 2007, 99: 202-211. DOI:10.1093/bja/aem133 |
| [6] |
陈本川. 减轻化学治疗引起的恶心与呕吐新药-盐酸罗拉吡坦)[J]. 医药导报, 2016, 35(5): 547-551. DOI:10.3870/j.issn.1004-0781.2016.05.033 |
| [7] |
PALIWALS, REICHARD G A, WANG C, et al. NK 1 antagonists: US, 8796299[P]. 2003-08-21.
|
| [8] |
WU G G, WERNG G, FU X Y. et al. Process and intermediates for the synthesis of 8-[{1-(3, 5-bis-(trifluoromethyl)phenyl)-ethoxy}-methyl]-8-phenyl-1, 7-diaza-spiro[4.5]decan-2-one compounds: WO, 2010028232[P]. 2010-03-11.
|
| [9] |
O′DONNELL M J, FANG Z Q, MA X J, et al. New methodology for the synthesis of α, α-dialkylamino acids using the "self-regeneration of stereocenters" method: α-ethyl-α-phenylglycine[J]. Heterocycle, 1997, 46: 617-630. DOI:10.3987/COM-97-S83 |
| [10] |
余长泉, 杨健, 许惠钢. 盐酸罗匹尼罗合成工艺的改进[J]. 高校化学工程学报, 2010, 24(6): 1011-1016. DOI:10.3969/j.issn.1003-9015.2010.06.017 |
| [11] |
YANG X H, XIE J H, LIU W P, et al. Catalytic asymmetric hydrogenation of δ-ketoesters: highly efficient approach to chiral 1, 5-diols[J]. Angew. Chem. Int. Edit., 2013, 52(30): 7833-7836. DOI:10.1002/anie.v52.30 |
| [12] |
REICHARD G A, HTENGONEH C, HPALIWALH S, et al. Asymmetric synthesis of 4, 4-disubstituted-2-imidazolidinones: potent NKR1R antagonists[J]. Org Lett, 2003, 23(5): 4249-4251. |