




2. 河南农业大学动物科技学院,郑州 450046;
3. 河南农业大学资源与环境学院,郑州450002
2. College of Animal Science and Technology, Henan Agricultural University, Zhengzhou 450046;
3. College of Resources and Environment, Henan Agricultural University, Zhengzhou 450002
秸秆焚烧和化肥不合理使用,使土壤微生物丰度和多样性降低,进而导致土壤酸化、板结、肥力下降等突出问题,不仅严重影响作物产量和粮食安全,而且对生态系统的可持续发展和绿色环境造成不利影响[1-3]。减少化肥使用,进行有机替代,增施生物肥料是解决土壤问题,实现农业绿色可持续发展的必然选择[4-6]。生物肥料是指以微生物及其代谢产物为核心,使农作物获得特定肥料效应的一类生物制品。生物肥料不仅为农作物提供丰富的碳、氮和矿物质等营养元素,更重要的是增加土壤中微生物的丰度和多样性以及酶活性,有效分解有机质,增强土壤肥力[7-8];生物肥是以畜禽粪便或作物秸秆为基质材料,经腐熟发酵而成,由于原料不同其微生物组成存在差异。研究表明畜禽粪肥施用在玉米上能够提高土壤生物活性,改善土壤理化性质,增加产量[9-10],但畜禽粪肥资源比较稀少,在玉米上很难承担成本费用。秸秆还田是当前华北地区秸秆处理的主要措施,秸秆不经腐熟发酵处理直接还田易受土壤微生物丰度、土壤温度、pH值和含水量等条件的制约,影响到秸秆的腐解和利用[11-12]。另外,秸秆连续还田后增加了土壤中的有机质,对土壤微生物多样性结构具有促进作用[13-14],但其机制和效果需要深入研究。植物根际微生物类似于动物的肠道微生物,对植物的生长和健康发挥重要作用,植物从土壤中获取各种营养物质往往是通过微生物代谢实现的。本研究以玉米根际微生物为对象,研究秸秆连续还田对根际微生物菌群结构的影响,探索秸秆还田替代畜禽粪肥的可行性。
根际微生物是指紧密附着于根际土壤颗粒中的微生物群,包括细菌和真菌,其中细菌占微生物总数的90%以上,它们作为一个复杂的生态体系,在调节陆地碳动态、养分循环和植物生产力方面发挥着关键作用。它们和植物间是互生关系,与植物根际相互作用、相互促进。在植物生长过程中,根际分泌的无机物和有机物为微生物提供了营养物质或改变了土壤环境条件,从而调节根际微生物的生长和结构组成[15-17]。微生物聚集在根际周围,由呼吸作用放出二氧化碳或代谢产酸有助于难溶矿物质的溶解,增加植物对磷及其他矿质元素的吸收;微生物合成多种蛋白酶,把有机物降解转化,为植物提供有效的养料[18];同时,微生物还能产生氨基酸、维生素、抗生素、生长刺激素(如吲哚乙酸,赤霉素等)等,抑制病原菌的繁殖,促进植物生长[19]。根际微生物作为土壤生态系统的重要指标,与作物产量和品质存在紧密相关性。因此,以根际微生物作为评价对象比单纯以营养元素含量为标准评价生物有机肥更为科学。
玉米(Zea mays L.)是世界上种植范围最广泛、土地利用率最高的粮食作物之一,在我国黄淮海地区种植面积最广。玉米相比小麦其生长期在夏季,气温高,土壤微生物繁殖旺盛,是研究根际微生物的适宜作物。近年来小麦和玉米秸秆连续还田已在华北地区广泛推广。研究表明秸秆连续还田能够增加根际微生物丰度和多样性[20],抑制土壤病原菌[21]。但是秸秆连续还田对玉米根际微生物菌群组成和多样性的影响与施用畜禽粪肥相比有何差异,该方面研究尚未见报道。本研究应用16S rDNA基因测序技术,研究秸秆连续还田对玉米根际微生物群落结构的影响,与施用畜禽粪肥相比较存在哪些差异,旨在为华北地区农作物多种有机替代提供依据。
1 材料与方法 1.1 材料试验用种子为郑原玉432玉米,由市场采购。
1.2 方法 1.2.1 试验地点试验于河南农业大学国家“2011计划”现代农业科技试验园区进行。试验设计为二组,分别为秸秆连续还田组和禽粪有机肥施用组,面积为50 m×200 m。大垄双行种植,垄上行距40 cm,垄间行距60 cm。
1.2.2 样品分组与处理秸秆还田组(Straw returning,SR)为小麦和玉米轮作,玉米秸秆翻耕时全量深埋还田,小麦秸秆粉碎全量覆盖还田,连续3年。施用畜禽粪肥(Livestock and poultry manure,LM)区块无秸秆还田,使用的畜禽粪肥是利用猪鸡粪便为主要原料经腐熟发酵而成。在小麦播种前土壤翻施畜禽粪肥100 kg/667 m2,在玉米播种后随灌溉时再次撒施100 kg/667 m2,全年分二次共施用200 kg/667 m2。玉米生育期按照高产田进行田间管理。玉米成熟期随机采样,分别标记为秸秆还田组(A1-6和B1-6)和畜禽粪肥组(C1-6和D1-6),AC和BD分别由不同的人采样。分别对两组玉米穗粒数、单粒重、百粒重等产量性状进行统计计算。
每个样品,以茎根10 cm半径范围,挖取0-40 cm深的根系,轻轻抖动,去掉根系上脱落的土壤,剪取根系并保留紧密附着在根系上的土壤,置于装有灭菌的0.86%NaCl溶液的50 mL离心管中,冰中放置30 min,每5 min摇匀一次,然后弃掉玉米根际,4 000 ×g、4℃离心30 min后倒掉上清液,所得的土壤沉淀物保存在无菌EP管中,置于-20℃备用。
1.2.3 土壤微生物DNA提取、PCR扩增及扩增产物的纯化回收应用土壤微生物DNA提取试剂盒(TIANGEN),按照使用说明提取样品DNA,应用Nanodrop对DNA进行定量后,通过1.2%琼脂糖凝胶电泳检测DNA的质量。以细菌总DNA为模板,应用通用引物515F(5'-gtgccagcccgg-3')和907R (5'-CC-GT-caattcmtragtt-3')扩增16S rDNA的V4-V5区,扩增体系为25 μL。扩增产物磁珠纯化回收并荧光定量。
1.2.4 制备测序文库和上机高通量测序采用Illumina公司的TruSeq Nano DNA LT Library Prep Kit并按照说明制备测序文库。上机测序前,采用Agilent High Sensitivity DNA Kit对文库进行质检,质检合格的文库采用Quant-iT PicoGreen dsDNA Assay Kit在Promega QuantiFluor荧光定量系统上对文库进行检测定量。根据操作说明使用Illumina MiSeq测序平台对文库进行双端测序。本研究中测序所获得的原始数据已存放在CNGBdb的CNSA(https://db.cngb.org/CNSA/)中,注册号为CNP0000832。
1.2.5 生物信息学分析首先对高通量测序的原始下机数据根据序列质量进行初步筛查;对问题样本进行重测、补测。通过质量初筛的原始序列按照index和Barcode信息,进行文库和样本划分,并去除barcode序列。按照QIIME2 dada2分析流程进行序列去噪、OTUs聚类。根据ASV/OTU在不同样本中的分布,评估每个样本的Alpha多样性水平,以Chao1指数反映丰富度,以Shannon和Simpson指数反映多样性,以Pielou值反映均匀度。在ASV/OTU层面,计算各样本的距离矩阵,并通过非约束排序,结合相应统计学检验方法,衡量不同样本(组)间的beta多样性差异及差异显著性,并使用PICRUSt方法,基于16S rDNA序列对微生物群的功能潜能进行预测[22]。
1.2.6 统计分析结果以“平均值±标准误(means±SE)”表示。数据采用SPSS 24.0中独立样本t检验进行显著性分析,P < 0.05表示显著差异,P < 0.01表示极显著差异。
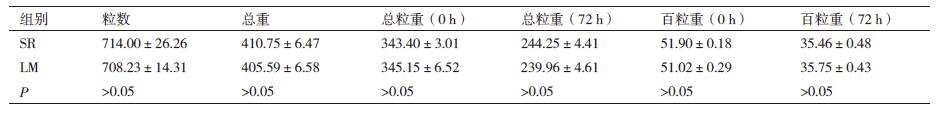
2 结果 2.1 玉米穗部性状统计结果两组玉米穗部性状统计结果(表 1)。SR组与LM组在粒数(粒)、总重(g)、总粒重(g)等产量相关的指标之间无显著差异(P > 0.05)。
利用高通量测序技术研究了不同处理方式的玉米根际菌群的多样性。通过序列比对和注释表明,在门水平上,两组玉米根际菌群中的大多数归属于30多个门水平,图 1(前20门)显示优势菌门为变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、绿弯菌门(Chloroflexi)和己科河菌门(Rokubacteria)等,在门水平上两组的主要菌门构成具有相似性。进一步分析发现,这两组微生物群中变形菌门(Proteobacteria)的比例较高,且变形菌门(Proteobacteria)均为优势菌门,但在LM组,变形菌门(Proteobacteria)的相对丰度显著高于SR组(P < 0.01)。SR组中厚壁菌门(Firmicutes)和绿弯菌门(Chloroflexi)的相对丰度显著高于LM组(P < 0.01)。

|
| 图 1 SR和LM两组玉米的根际微生物在门分类水平的菌群组成 |
在属水平,两组玉米的根际微生物覆盖了100多个类群。图 2(前20属)显示了这两个类群的菌群结构和相对丰度。优势菌属主要包括芽胞杆菌属(Bacillus sp.)、根瘤菌属(Rhizobium)、黄杆菌属(Flavobacterium)、硝化螺旋菌属(Nitrospira)、假单胞菌属(Pseudomonas)、诺卡菌属(Nocardioides)、壤霉菌属(Agromyces)和Burkholderia等。在属水平上两组的主要菌属构成具有相似性。但芽孢杆菌属在SR组中显著高于LM组(P < 0.01),而壤霉菌属(Agromyces)在LM组中显著高于SR组。

|
| 图 2 SR和LM两组玉米的根际微生物在属分类水平的菌群组成 |
Alpha多样性是指特定区域或生态系统的多样性。多样性指数包括Chao1指数、ACE指数、Simpson指数和Shannon指数,它们反映了样品的丰富度、多样性和均匀度等方面的指标。Chao1或ACE指数越大,群落的丰富度就越大。表 2显示SR组与LM组的Alpha多样性指数均无显著差异(P > 0.05),说明两组玉米的根际微生物群的多样性和丰富度相似,具有较好的稳定性。

Beta多样性指数聚焦于不同生态系统之间多样性的比较,表示样本间的差异。主坐标分析(Principal coordinates analysis,PCoA)便是一种最经典的分析样本间差异的分析方法。它通过将样本距离矩阵经过投影后,在低维度空间进行展开,并最大限度地保留原始样本的距离关系。PCoA以样本距离为整体考虑,相比于主成分分析(Principal analysis,PCA),更符合生态学数据特征,因此应用PCoA比较样本间的差异。在主坐标分析图中(图 3),两组处理的玉米根际菌群组内差异较小,群落组成更为相似,而组间比较表明两组处理玉米根际菌群群落组成差异较大,表明玉米根际菌群结构显著受粪源微生物和有机质差异的影响。

|
| 图 3 SR和LM两组玉米的根际微生物的PCoA结果 |
为了进一步比较样本间的物种组成差异,实现对各样本的物种丰度分布趋势的展示,应用热图对SR和LM两组玉米根际微生物群进行物种组成分析。平均丰度前50位的属的丰度数据被绘制热图(图 4)。在对样本聚类的图中,样本按照物种组成数据的欧式距离(Euclidean distance)进行UPGMA聚类,并根据聚类结果排列。在对物种聚类的图中,按照其组成数据的Pearson相关性系数矩阵进行UPGMA聚类,并根据聚类结果或者物种在样本中的平均丰度排序。图中,红色色块代表该属在该样本中的丰度较其他样本高,蓝色色块代表该属在该样本中的丰度较其他样本低。结果显示A、B之间和C、D之间菌群的相对丰度无显著差异(P > 0.05),表明不同人员采样对结果的影响较小。但是SR组(A组+B组)和LM组(C组+D组)之间存在较大差异(P < 0.05),与Beta多样性分析结果一致,表明受粪源微生物和有机质差异的影响较为明显。其中SR组的土壤单胞菌属(Gemmatimonas)、硝化螺旋菌属(Nitrospira)、生丝微菌属(Hyphomicrobium)、芽孢杆菌属(Bacillus)等的相对丰度高于LM组,而LM组的乳酸杆菌属(Lactobacillus)、鞘脂单胞菌属(Sphingomonas)、土壤杆菌属(Pedobacter)、壤霉菌属(Agromyces)等的相对丰度显著高于SR组(P < 0.05)。

|
| 图 4 SR和LM两组根系微生物群的物种聚类在属水平的组成热图 |
KEGG数据库的核心为生物代谢通路分析数据库(KEGG Pathway Database,(http://www.genome.jp/kegg/pathway.html),分为6大类,包括代谢(Metabolism)、遗传信息处理(Genetic Information Processing)、环境信息处理(Environmental Information Processing)、细胞进程(Cellular Processes)、生物体系统(Organismal Systems)和人类疾病(Human Diseases)。在本研究中,两组处理的玉米根际菌群的KEGG功能通路的丰度结果见图 5。结果显示,SR组和LM组的玉米根际微生物共参与KEGG的6大类功能通路,包括45种子功能通路,且大多数富集于代谢通路,包括碳水化合物代谢、氨基酸代谢、辅酶和维生素的代谢、萜类化合物和聚酮类化合物代谢和异源物质生物降解和代谢等。尽管在Beta多样性分析中两组菌群构成存在显著差异,但图 6显示SR组与LM组主要功能基因分布无显著差异,表明不同微生物在功能上可能存在互补性。

|
| 图 5 SR和LM两组玉米的根际微生物群的功能潜能预测 |

|
| 图 6 SR和LM两组玉米的根际微生物群代谢功能基因分布 |
本研究应用16S rDNA高通量测序技术对玉米根际微生物菌群进行分析。结果显示玉米根际微生物主要群落构成在门水平和属水平上两组具有相似性,且两组共有的OTUs达到7 849,独有的OUTs分别为12 800(SR组)和11 297(LM组)。24个样本共覆盖30余个细菌类群,其中变形菌门、放线菌门、酸杆菌门、拟杆菌门、厚壁菌门、绿弯菌门和棒状杆菌是玉米根际中的优势菌门。在属水平,优势菌属主要包括芽胞杆菌属、根瘤菌属、黄杆菌属、硝化螺旋菌属、假单胞菌属、诺卡菌属、壤霉菌属和Burkholderia等。在本研究中两组均具有较高的微生物丰度,但玉米产量差异不显著,此结果与Ma等[23]的报道相一致。杨志臣等[24]研究显示直接施用作物秸秆同施用腐熟有机肥对土壤的培肥效果基本相同,均对土壤理化性质有很大的改善,因此秸秆直接还田与施用畜禽粪肥具有同等的培肥土壤的效果。在本研究中,LM组的变形菌门的相对丰度要明显高于SR组,但SR组中厚壁菌门的相对丰度要明显高于LM组。在属水平上,SR组的芽胞杆菌属的相对丰度要明显高于LM组,而壤霉菌属的相对丰度低于LM组。粪源微生物组成和有机质可利用碳源的差异可能是影响土壤细菌多样性与群落结构差异的重要因素[25-26]。秸秆主要由纤维素、半纤维素和木质素组成,芽胞杆菌能够利用纤维素和半纤维素作为生长的碳源。而在猪、鸡为主的畜禽粪肥中秸秆碳源含量相对较低,可能是芽胞杆菌属在LM组中占比低于SR组的原因之一[27]。根际微生物与植物根系之间具有互作关系,根际微生物生长繁殖为植物生长提供了营养元素,同时植物根系也为微生物的生长提供了生存环境,间接选择和促进了“适者生存”的根际微生物[28]。秸秆还田和畜禽粪肥两种处理的玉米根际微生物主要菌群构成具有相似性,这一结果是否与玉米品种相关尚需进一步研究[29]。
玉米根际微生物群落多样性指数反映了玉米根际微生物群落结构的复杂程度。通常来讲,群落Alpha多样性指数(Chao1指数、ACE指数、Simpson指数和Shannon指数)越高,表明微生物群落结构越复杂,群落的稳定性就越好。在本研究中,SR组与LM组的Alpha多样性指数无显著差异,表明两组玉米根际微生物群落均具有较高的丰富度、多样性和稳定性。两组玉米根系微生物中包含许多益生菌如枯草芽胞杆菌、乳酸杆菌和解淀粉芽胞杆菌,这些微生物具有较强的益生功能,其不但能够抑制植物病原菌,而且还能够激发植物自身免疫力,从而增强植物的抗病性能[30-33]。菌群代谢功能预测的结果显示SR组和LM组的玉米根际微生物参与代谢通路,主要包括氨基酸代谢、碳水化合物代谢、辅酶和维生素的代谢和萜类化合物和聚酮类化合物的代谢。尽管在门水平和属水平上两组中有部分菌群含量存在差异,但在功能预测分析上两组的差异不明显,表明不同微生物之间存在着功能多样性,不同的玉米根系微生物通过相似的功能通路各自发挥着重要作用[34-35]。
4 结论本研究利用高通量测序技术对不同处理方式的玉米根际微生物的多样性进行测序分析,发现不同处理方式的玉米根际微生物的构成和多样性具有相似性,两者的产量无显著差异。表明秸秆连续还田与畜禽粪肥施用具有同样的效果,都能提升玉米根际微生物丰度和多样性。
| [1] |
Kavamura VN, Rifat H, Clark IM, et al. Inorganic nitrogen application affects both taxonomical and predicted functional structure of wheat rhizosphere bacterial communities[J]. Front Microbiol, 2018, 9(5): 1074. |
| [2] |
Wang Q, Jiang X, Guan D, et al. Long-term fertilization changes bacterial diversity and bacterial communities in the maize rhizosphere of Chinese Mollisols[J]. Applied Soil Ecology, 2018, 125: 88-96. DOI:10.1016/j.apsoil.2017.12.007 |
| [3] |
Li M, Wang T, Li L, et al. Effects of long-term nitrogen fertilizer application on rhizosphere microorganisms under different soil types[J]. Pol J Environ Stud, 2019, 28(3): 1771-1784. DOI:10.15244/pjoes/89583 |
| [4] |
Bhardwaj D, Ansari M, Sahoo R, et al. Biofertilizers function as key player in sustainable agriculture by improving soil fertility, plant tolerance and crop productivity[J]. Microbial Cell Factories, 2014, 13(1): 66. DOI:10.1186/1475-2859-13-66 |
| [5] |
Mahanty T, Bhattacharjee S, Goswami M, et al. Biofertilizers :a potential approach for sustainable agriculture development[J]. Environ Sci & Pollu Res, 2017, 24(4): 3315-3335. |
| [6] |
Aloo BN, Makumba BA, and Mbega ER. The potential of bacilli rhizobacteria for sustainable crop production and environmental sustainability[J]. Microb Res, 2018, 219(2): 26-39. |
| [7] |
Xu H, Shao H, Lu Y. Arbuscular mycorrhiza fungi and related soil microbial activity drive carbon mineralization in the maize rhizosphere[J]. Ecotoxi Environ Safety, 2019, 182(7): 109476. |
| [8] |
Kheirfam H, Sadeghi SH, Homaee M, et al. Quality improvement of an erosion-prone soil through microbial enrichment[J]. Soil and Tillage Research, 2017, 165: 230-238. DOI:10.1016/j.still.2016.08.021 |
| [9] |
Vollú RE, Cotta SR, et al. Response of the bacterial communities associated with maize rhizosphere to poultry litter as an organomineral fertilizer[J]. Front Environ Sci, 2018, 6: 118. DOI:10.3389/fenvs.2018.00118 |
| [10] |
Lu H, Lashari MS, et al. Changes in soil microbial community structure and enzyme activity with amendment of biochar-manure compost and pyroligneous solution in a saline soil from Central China[J]. Europ J Soil Biol, 2015, 70: 67-76. DOI:10.1016/j.ejsobi.2015.07.005 |
| [11] |
Yan C, Yan S, Jia T, et al. Decomposition characteristics of rice straw returned to the soil in northeast China[J]. Nutrient Cycling in Agroecosystems, 2019, 114(3): 211-224. DOI:10.1007/s10705-019-09999-8 |
| [12] |
Zhang K, Chen L, Li Y, et al. Interactive effects of soil pH and substrate quality on microbial utilization[J]. Europ J Soil Biol, 2020, 96: 103151. DOI:10.1016/j.ejsobi.2020.103151 |
| [13] |
Chen Y, Xin L, Liu J, et al. Changes in bacterial community of soil induced by long-term straw returning[J]. Scientia Agricola, 2017, 74(5): 349-356. DOI:10.1590/1678-992x-2016-0025 |
| [14] |
Qiao Y, Miao S, Zhong X, et al. The greatest potential benefit of biochar return on bacterial community structure among three maizestraw products after eight-year field experiment in Mollisols[J]. Applied Soil Ecology, 2019, 147: 103432. |
| [15] |
Jin Y, Zhu H, Luo S, et al. Role of maize root exudates in promotion of colonization of bacillus velezensis strain S3-1 in rhizosphere soil and root tissue[J]. Curr Microbiol, 2019, 76(5): 855-862. |
| [16] |
Kong X, Han Z, Tai X, et al. Maize(Zea mays L.)varieties significantly influence bacterial and fungal community in bulk soil, rhizosphere soil, and phyllosphere[J]. FEMS Microbiology Ecology, 2020, 96(3): 3. |
| [17] |
Huang X, Chaparro JM, Reardon KF, et al. Rhizosphere interactions :root exudates, microbes, and microbial communities[J]. Botany, 2014, 92(4): 267-275. DOI:10.1139/cjb-2013-0225 |
| [18] |
Huang R, Zhang Z, Xiao X, et al. Structural changes of soil organic matter and the linkage to rhizosphere bacterial communities with biochar amendment in manure fertilized soils[J]. Science of the Total Environment, 2019, 692(11): 333-343. |
| [19] |
Re nu, Gupta SK, Rai AK, et al. Metaproteomic data of maize rhizosphere for deciphering functional diversity[J]. Data in Brief, 2019, 27(9): 104574. |
| [20] |
Zhang M, Yi L, Rong S, et al. Effects of rice straw returning on the community structure and diversity of nitrogen-fixing gene(nifH) in paddy soil[J]. J Appl Ecol, 2013, 24(8): 2339-2344. |
| [21] |
Yang H, Ma J, Rong Z, et al. Wheat straw return influences nitrogen-cycling and pathogen associated soil microbiota in a wheat-soybean rotation system[J]. Front Microbiol, 2019, 10(8): 1811. |
| [22] |
Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences[J]. Nature Biotechnology, 2013, 31(9): 814-921. DOI:10.1038/nbt.2676 |
| [23] |
Ma Z, Xie Y, Zhu L, et al. Which of soil microbes is in positive correlation to yields of maize(Zea mays L[J]. Plant Soil and Environment, 2017, 63(12): 574-580. DOI:10.17221/590/2017-PSE |
| [24] |
杨志臣, 吕贻忠, 张凤荣, 等. 秸秆还田和腐熟有机肥对水稻土培肥效果对比分析[J]. 农业工程学报, 2008, 24(3): 214-218. Yang ZZ, Lv YZ, Zhang FR, et al. Comparative analysis of the effects of straw-returning and decomposed manure on paddy soil fertility betterment[J]. Transactions of the Chinese Society of Agricultural, 2008, 24(3): 214-218. |
| [25] |
Yu C, Li Y, Mo R, et al. Effects of long-term straw retention on soil microorganisms under a rice-wheat cropping system[J]. Archives of Microbiology, 2020, 202(7): 1915-1927. DOI:10.1007/s00203-020-01899-8 |
| [26] |
Rieke EL, Soupir ML, Moorman TB, et al. Temporal dynamics of bacterial communities in soil and leachate water after swine manure application[J]. Front Microbiol, 2018, 9: 3197. DOI:10.3389/fmicb.2018.03197 |
| [27] |
Zhang L, Li L, Pan X, et al. Enhanced growth and activities of the dominant functional microbiota of chicken manure composts in the presence of maize straw[J]. Front Microbiol, 2018, 9: 1131. DOI:10.3389/fmicb.2018.01131 |
| [28] |
Jacoby RP, Kopriva S. Metabolic niches in the rhizosphere microbiome :New tools and approaches to analyse metabolic mechanisms of plant-microbe nutrient exchange[J]. J Exp Bot, 2018, 70(4): 1087-1094. |
| [29] |
Aira M, Gómez-Brandón M, Lazcano C, et al. Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities[J]. Soil Biology and Biochemistry, 2010, 42(12): 2276-2281. DOI:10.1016/j.soilbio.2010.08.029 |
| [30] |
Hu J, Wei Z, Kowalchuk GA, et al. Rhizosphere microbiome functional diversity and pathogen invasion resistance build up during plant development[J]. Environmental Microbiology, 2020. DOI:10.1111/1462-2920.15097 |
| [31] |
Pieterse CMJ, Zamioudis C, Berendsen RL, et al. Induced systemic resistance by beneficial microbes[J]. Annual Review of Phytopathology, 2014, 52(1): 347-375. DOI:10.1146/annurev-phyto-082712-102340 |
| [32] |
Ongena M, Jacques P, Touré Y, et al. Involvement of fengycin-type lipopeptides in the multifaceted biocontrol potential of Bacillus subtilis[J]. Appl Microbi Biotech, 2005, 69(1): 29-38. |
| [33] |
Wu Y, Zhou J, Li C, et al. Antifungal and plant growth promotion activity of volatile organic compounds produced by Bacillus amyloliquefaciens[J]. MicrobiologyOpen, 2019, 8(8). |
| [34] |
Zhou Y, Coventry DR, Gupta VVSR, et al. The preceding root system drives the composition and function of the rhizosphere microbiome[J]. Genome Biol, 2020, 21(1): 89. |
| [35] |
Qu Q, Zhang Z, Peijnenburg WJGM, et al. Rhizosphere microbiome assemble and its impact on plant growth[J]. Journal of Agricultural and Food Chemistry, 2020, 68: 5024-5038. DOI:10.1021/acs.jafc.0c00073 |