




2. 中国农业科学院深圳农业基因组研究所,深圳 518120
2. Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518120
基因组Fosmid文库技术自1992年被创建之后,即广泛地应用于基因组学研究当中[1-12]。1996年,Preston等[13]利用Fosmid文库识别并鉴定了寄生在海绵中的一种嗜冷泉古菌(Cenarchaeum symbiosum)。1997年,Fitz-Gibbon等[14]利用Fosmid文库构建了一种超嗜热古细菌(Pyrobaculum aerophilum)的物理图谱,并预测了其基因组含474个基因。1998年,Deckert等[15]用“鸟枪”测序法破解超嗜热菌(Aquifex aeolicus)的基因组时,借助Fosmid文库来填补重叠群之间的空缺。1999年,Cui等[16]构建两个自交不亲和油菜株系的Fosmid文库,通过对文库中的部分克隆测序,对控制芸苔属植物的自交不亲和性的S位点区间进行了结构和转录分析。2004年,国际人类基因组测序联合会公布的已完成测序的人类基因组有341个空缺,包括位于常染色质区域的250个空缺,Bovee等[17]利用Fosmid文库将常染色质区域的26个空缺补全,67个空缺平均每个都填补约32 kb。
新一代测序技术,尤其是读长可达20 kb甚至40 kb以上的单分子测序技术的不断进步,使Fosmid文库的应用日渐式微。但是,Fosmid文库在当前的基因组学研究中仍然具有其独特的价值。第一,与细菌人工染色体(Bacterial artificial chromosome,BAC)文库类似,Fosmid文库可以用于基因图位克隆过程中目标区域序列的精确测序。目前,虽然有很多物种都已经有了参考基因组序列,但同一物种不同品种之间的基因组序列通常存在很大差异[18]。这导致在图位克隆基因的过程中,将基因精细定位后,有时难以根据参考基因组序列获得目标区段的准确序列。此时,需要构建BAC或Fosmid文库,并从中筛选位于目标区域的克隆进行测序,从而获得候选基因的准确序列。例如,2016年,Zhao等[19]在通过图位克隆法鉴定水稻褐飞虱抗性基因BPH9时,利用Fosmid文库确定了抗性亲本Pokkali定位区间的68 kb DNA序列,从而确认了候选基因并获得其准确序列,这样的例子还有很多,如小麦条锈病抗性基因Yr28的克隆等[20]。第二,虽然测序技术已经取得了相当大的发展,但在全基因组测序的过程中,仍然经常使用Fosmid文库辅助组装或者验证基因组组装的准确度。例如,2014年,松树和甜菜的基因组组装研究者都借助了Fosmid文库[21-22]。2017年,在水稻蜀恢498基因组的组装过程中,研究者利用来自564个Fosmid混合池(池容量为1 000-1 500个单克隆)的简化基因组测序数据,将来自于全基因组的PacBio reads互相区分并分别组装,用于降低基因组组装的复杂度,开创了Fosmid文库在基因组组装中的应用新方向[18]。此外,Fosmid文库还被用于人类基因组的单倍型组装[23]。2019年,在糜子的全基因组测序中,研究者利用Fosmid文库单克隆测序验证基因组组装的准确度[24]。第三,在宏基因组学的研究中,Fosmid文库也可以被用来保存环境样本中所包含的全部微生物的基因组DNA,并用于后续的基因功能分析[25-26]。
Fosmid载体由cosmid载体发展而来,最初是Kim等[27]将带有大肠杆菌致育(Fertility,F)因子的pBAC载体与Cosmid载体pUCcos融合后构建成pFOS1载体。Cosmid载体是一种最老的高容量载体,可插入28-45 kb的外源DNA;应用Cosmid载体构建基因组文库时,选择28-45 kb的DNA片段与载体连接,可保证包装后的DNA高效进入λ噬菌体头部,从而提高建库效率[28]。在连接过程中,样品的DNA小片段之间可能相互连接,之后再与载体连接,从而产生包含来自两个或两个以上不相邻基因组区段的嵌合克隆[28]。另外,由于大肠杆菌携带的重组子拷贝数很高,黏粒中克隆的DNA会发生重排[28]。Kim等[27]将F因子引入Cosmid载体构成Fosmid载体,保证了构建的重组质粒DNA在每个细胞中只有1-2个分子,从而极大降低了克隆中外源DNA的重排水平。若要减少Fosmid文库中嵌合克隆的数量,可在建库时尽量降低连接反应中小片段DNA的含量。因此,对外源DNA片段大小进行准确地筛选,可排除小片段DNA从而减少嵌合克隆;同时,排除超过45 kb的大片段DNA,可以提高文库的制备效率。在Fosmid文库构建试剂盒(Epicentre,美国)的使用说明中,建议外源DNA片段大小为40 kb左右。但是,从琼脂糖凝胶中回收、纯化长度超过20 kb的DNA片段是分子生物学实验的一个难点,且长度越长,回收效率越低。因此,准确、高效的回收40 kb的DNA片段是Fosmid文库构建的一个重要且难度较高的步骤。
当前,虽然有多种方法可以用于分离40 kb范围内的DNA片段,包括磁珠、硅胶膜(Silica membrane)或硅胶颗粒(Silica particles)以及密度梯度离心,但是它们的回收效率都较低。综合比较不同方法,传统的切胶并电洗脱至透析袋,仍然是回收40 kb DNA的最好方法[28]。2015年,Sage Science公司开发出了SageELF仪器,将传统的切胶并利用透析袋回收DNA的过程自动化,不仅简化了利用透析袋回收DNA的操作流程,也提高了DNA的回收效率。目前,在新一代测序领域,Sage ELF已经被广泛应用于不同长度的DNA片段回收[29],如100 bp的miRNA文库、400 bp左右的Truseq文库、10 kb左右的Mate pair文库和1-5 kb的PacBio iso-Seq文库。但是,Sage ELF仪器中并未设置回收40 kb的方法。
本研究利用SageELF提供的0.75%琼脂糖胶盒,尝试将其内置的回收10 kb DNA的程序,改造成回收40 kb DNA的程序。我们对这一方法进行了测试,并成功建立了高效的40 kb DNA回收方法;同时,利用改良的方法回收到的DNA样品构建了Fosmid文库,并对文库中插入的外源DNA片段大小进行评估,结果显示:高效、准确的40 kb DNA回收方法大大降低了Fosmid文库构建的难度。综上所述,本研究对Fosmid文库构建过程中,样品DNA片段的制备方法进行改良,旨在为科学构建Fosmid文库提供改进方向和实验证据,为生物的基因组学研究奠定基础。
1 材料与方法 1.1 材料Fosmid载体质粒pFosill-2(Genbank登录号JX069762)[30],由美国Broad研究所Andreas Gnirke博士惠赠;所用籼型水稻品种是华占。
1.2 方法 1.2.1 基因组DNA提取水稻华占基因组DNA,用CTAB法提取[31]。
1.2.2 基因组DNA的定量与打断利用Qubit 3.0荧光计(Invitrogen,美国)测定水稻基因组DNA浓度,用无菌双蒸水将浓度调至200 ng/µL;吸取40 µL DNA溶液加入到g-TUBE(Covaris,美国)中,5 000 r/min离心3 min后,将g-TUBE倒转放入离心机,5 000 r/min离心3 min,上述步骤重复5次后,吸出打断后的DNA样品于新离心管中备用。重复上述步骤,直至得到足够量的打断DNA,然后利用脉冲电泳检测打断DNA的片段分布,确保主要片段集中在40 kb左右。
1.2.3 基因组DNA片段的分选与回收打断后的DNA加入Loading buffer备用,使用全自动核酸/蛋白质回收仪SageELF(Sage science,美国)进行DNA片段的分选与回收。选用0.75%琼脂糖胶盒(Agarose Gel cassette,Sage science,美国),根据操作说明进行并有改动。主要步骤如下:排除胶盒气泡并清洗胶盒上的回收孔,然后将胶盒置于仪器上的盒槽中并设置恰当的缓冲液水平。电流测试通过后,在胶盒的点样孔中加入准备好的DNA样品,注意上样量不超过60 µL,选用“时间”模式,电泳10 h,为了避免长时间电泳过程中电泳液的蒸发,每隔3 h暂停一次,补水约3-5 mL并混匀。电泳结束后,收集每个回收孔中的DNA于一新离心管中,用Qubit 3.0测定每孔回收到的DNA浓度,回收的DNA可短期保存于4 ℃。
1.2.4 脉冲场凝胶电泳(Pulse-Field Gel Electropho-resis,PFGE)检测DNA片段大小利用SeaKem® Gold Agarose(Lonza,德国)及0.5×TBE电泳缓冲液制备1%的琼脂糖凝胶,使用脉冲场凝胶电泳仪(Rotaphor 6.0,Analytik Jena,德国)检测DNA片段大小,操作按照仪器说明书进行,电泳条件如下:电压180 V,脉冲夹角120°,间隔时间60 s,温度13 ℃,电泳时间24 h。电泳结束后,将胶块放入GelRed(Vazyme,南京)溶液中染色约30 min,蒸馏水脱色约10 min,然后用凝胶成像系统(GelDoc XR+,BioRad,美国)拍摄图像。
1.2.5 Fosmid文库的构建首先,对载体质粒pFo-sill-2线性化与去磷酸化:取200 µg pFosill-2 DNA,使用Plasmid-SafeTM ATP-Dependent Dnase(Epicentre,美国)进行消化以去除载体质粒提取过程中发生断裂的DNA,使用0.8×磁珠(VAHTS DNA Clean Beads,Vazyme,南京)纯化出环状载体质粒DNA后,利用AatⅡ(Fermentas,美国)及Eco72Ⅰ(Fermentas,美国)对其双酶切以获得两端的载体臂,0.8×磁珠纯化酶切产物,接着使用Calf Intestinal Alkaline Phosphatase(NEB,英国)进行去磷酸化,最后用酚/氯仿抽提两次纯化载体臂。
接着,利用KAPA Library Preparation Kit(KAPA biosystems,美国)对基因组DNA片段进行末端补平,0.8×磁珠纯化补平产物后,利用KAPA T4 DNA Ligase(KAPA biosystems,美国)连接载体臂与基因组DNA片段,室温连接1 h后,70 ℃处理10 min终止反应。然后,使用MaxPlaxTM Lambda噬菌体包装提取物(Epicentre,美国)对连接产物进行体外包装,包装产物侵染大肠杆菌菌株DH10T,然后将侵染反应液涂布于含12.5 µg/mL氯霉素的固体LB培养基表面,置于37 ℃培养箱,倒置培养过夜。同时,将包装产物分别稀释10倍、100倍、1 000倍3个梯度侵染DH10T后培养,用于文库滴度检测。
1.2.6 文库重组克隆的分析从上一步的培养皿中,随机挑选10个单克隆,分别接种于50 mL LB液体培养基(包含34 µg/mL氯霉素)摇培过夜,使用碱裂解法提取重组质粒后,酶切质粒DNA检测阳性率。随后,又随机挑选25个单克隆,分别接种于1 mL LB液体培养基摇培约15 h后,分成两份:一份送苏州金唯智生物科技有限公司或者生工生物工程(上海)股份有限公司,利用pFosill-2载体插入位点两侧的T7和SP6启动子位点对每个重组质粒的两端进行Sanger测序,获得的序列与蜀恢498的基因组序列(http://www.mbkbase.org/)进行Blast分析,从而获知插入片段的大小;另一份进行扩大培养并提取重组质粒,每个重组质粒取1 µg使用Not Ⅰ(Thermo Fisher,美国)37 ℃酶切3 h,随后PFGE检测(电压130 V,脉冲夹角120°,间隔时间4 s,温度10 ℃,电泳时间18 h)载体带以及外源基因组DNA片段的酶切带型。最后,根据末端测序的结果下载蜀恢498的基因组序列,利用CodonCode Aligner软件(CodonCode Corporation,美国)进行电子酶切模拟,得到虚拟酶切图,与PFGE电泳图进行对比。
2 结果 2.1 改进的Sage ELF操作流程可准确、高效地回收40 kb左右的DNA片段为回收40 kb的DNA片段,将提取的基因组DNA经g-TUBE打断后,取20 µg利用SageELF仪器进行DNA片段的分选与回收。每个回收孔的DNA,均取50-100 ng进行PFGE电泳检测。通过测试不同的SageELF电泳时间发现,10 h的电泳可以有效地将10-50 kb的基因组DNA分开,此时,40 kb左右的DNA片段位于胶盒的第1-3孔,虽然这3个孔的DNA呈弥散分布,但仍可看出主带在40 kb附近,甚至有少量DNA片段稍大于48.5 kb(图 1),这部分DNA可用来构建Fosmid文库。此后的回收孔中,DNA片段大小依次降低且没有呈弥散分布,第4孔的DNA主带大小约30 kb,到最后一孔即第13孔,DNA主带大小在12 kb-15 kb之间(图 1)。

|
| M:DNA标记(CHEF DNA size standards 8-48 kb ladder,BioRad,美国);1-13:从胶盒第1至第13回收孔中回收到的DNA片段 图 1 Sage ELF回收的DNA片段的PFGE分析 |
利用Qubit 3.0测定每个孔回收到的DNA浓度发现,第1孔至第13孔依次回收得到275 ng、2 266 ng、4 576 ng、1 452ng、831 ng、649 ng、466 ng、451 ng、329 ng、311 ng、119 ng、243 ng DNA。打断的DNA片段大小峰值在40 kb。因此,回收量最高的DNA片段也集中在40 kb附近,这与预期的结果一致。以上DNA长度和浓度检测说明,利用改进的SageELF的电泳回收程序,可以高效地回收长度达40 kb的DNA片段,且操作流程简单易掌握。高浓度40 kb DNA片段的获得为Fosmid文库的构建奠定了坚实的基础。
2.2 Fosmid文库滴度及重组质粒的插入片段大小分析为检测文库容量以及覆盖率,包装产物用Phage Dilution Buffer稀释10倍、100倍、1 000倍3个梯度,混匀后分别取20 µL加入200 μL的大肠杆菌DH10T细胞中,37 ℃孵育30 min后涂氯霉素平板,倒置于37 ℃培养过夜。稀释100倍的平板上,平均每个平板长出12个单克隆,那么文库的总滴度约为12×100×1 000 / 20 = 6×104 CFU/mL。
由于28-45 kb的外源DNA片段包装后可高效进入λ噬菌体头部,本研究将第1至3孔的DNA混合,按照Williams等[30]的方法成功地构建了Fosmid文库。为了初步检测文库质量,随机挑选文库的10个单克隆,提取少量质粒DNA,用限制性内切酶Aat Ⅱ和Eco72Ⅰ作双酶切后进行PFGE分析发现,10个质粒均有外源DNA插入,文库阳性率为100%。但是,质粒的酶切产物条带过多,难以推测准确的外源DNA长度(数据未展示)。为了检测文库中外源DNA片段的准确大小,我们又随机挑选了25个单克隆进行双末端Sanger测序并成功获得了24个克隆的双末端序列,这些序列与蜀恢498的基因组序列[18]比对结果如表 1所示,插入的外源DNA片段大小在23 kb-52 kb之间,每个克隆的平均插入片段大小约为37.9 kb,标准差为5.2 kb,片段大小比较集中。
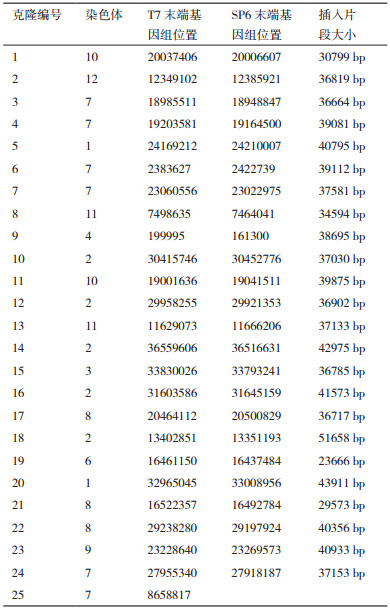
为了进一步确认测序克隆中插入的外源DNA片段大小,对表 1中的25个克隆,提取质粒,用Not Ⅰ酶切(可以切出完整的载体片段)后,进行PFGE检测。如图 2-A所示,酶切后的每个质粒都包含一条约7 kb的载体带(箭头所指)。为了便于分析外源DNA片段的酶切带型,我们根据表 1中蜀恢498的基因组位置信息,将对应的DNA序列提取出来,然后分析其中的Not Ⅰ识别位点的位置和数量,最后根据此结果预测外源片段的酶切带型(图 2-B)。结果发现,除6号、9号、11号、12号、14号和20号克隆外,其他18个克隆的酶切带型都与预测的带型一致,进一步验证了外源DNA片段的大小。

Fosmid文库在基因克隆、物理图谱构建及基因组测序等工作中被广泛应用[1-12]。最近几年,Fosmid文库与高通量测序技术相结合,如在人类单倍型测序[23, 30]、辅助基因组de novo组装[18]及宏基因组学研究[25-26]等方面,发挥了其独特的作用,使Fosmid文库在基因组学研究中的地位更加重要。但常规的Fosmid文库构建过程,尤其是40 kb DNA片段的回收,操作难度很大,限制了Fosmid文库构建的成功率,因此改良Fosmid文库的构建方法很有必要。
自Kim等[27]于1992年创建第一个Fosmid文库以来,构建Fosmid文库的方法就一直被改进。但目前看来,改进Fosmid载体的情况较多,外源基因组DNA片段的分选和回收方法,还是依赖传统的切胶并用透析袋回收或者密度梯度离心回收。切胶回收DNA,需经过PFGE、切胶、透析袋回收3步,流程比较麻烦;而密度梯度离心回收DNA,需经过超速离心、穿刺取样、透析纯化等步骤,流程更加繁琐。这两种方法,实验操作难度很大,技巧性也高,研究者往往难以快速掌握,且这些方法的DNA回收率也很低。利用我们改进的方法,只需将打断后的基因组DNA加到SageELF胶盒点样孔中,中间步骤完全由机器操作,最后从胶盒回收孔中吸取不同区段的DNA即可直接进行后续的分子生物学操作,流程简单、便捷。
全自动核酸/蛋白质回收系统SageELF是Sage Science公司2015年推出的仪器,将传统的切胶并利用透析袋回收DNA的过程自动化,不仅简化了利用透析袋回收DNA的操作流程,也提高了DNA的回收效率。目前,SageELF被广泛应用于新一代测序领域中,小至100 bp、大至20 kb左右的不同长度的DNA片段回收[29]。但是,SageELF仪器的中没有设置回收40 kb的方法,显然,Fosmid文库的构建,不是厂家考虑到的应用。与常规的Biorad的箝位匀强电场系统(CHEF)不同,SageELF使用的是横向交变电泳(TAFE),其电泳液的体积只有大约20 mL,如果用于40 kb片段的分离,需要长时间、高电流电泳。但是,在如此小的体积中长时间进行高电流电泳,极易导致电泳液温度过高和蒸发,因此,我们做了一系列的电流、脉冲时间的测试,并调整了仪器相应的运行监控参数,最终成功的从20 μg的input DNA中回收到约8 μg的40 kb左右的DNA。而实际上,使用Epicentre和Lucigen等公司的试剂盒,只需要100 ng-1 μg的40 kb DNA用于Fosmid文库构建。因此,我们建立的方法,可以很容易地回收到足够的40 kb DNA片段,大大降低了Fosmid文库构建的难度。
4 结论本研究通过改进全自动核酸/蛋白质回收系统SageELF的操作流程,实现了40 kb DNA片段的高效回收。利用我们改良的SageELF法回收40 kb左右的基因组DNA大片段,操作便捷,回收率高,片段大小精准,且文库插入片段集中,显著提高了Fosmid文库的构建质量。
| [1] |
Dai Z, Li T, Li J, et al. High-throughput long paired-end sequencing of a Fosmid library by PacBio[J]. Plant Methods, 2019, 15: 142. DOI:10.1186/s13007-019-0525-6 |
| [2] |
丁小维, 田文婷, 张波, 等. 盐湖微生物Fosmid文库纤维素酶克隆的筛选及产酶研究[J]. 陕西理工学院学报:自然科学版, 2016, 32: 59-63. |
| [3] |
董亮, 俞勤丽, 董志扬, 等. 沤麻废水处理池微生物多样性分析及其Fosmid文库酶活初步筛选[J]. 食品工业科技, 2015, 36: 167-172. |
| [4] |
窦怀乾, 李仰平, 吕佳, 等. 基于WGP物理作图法的虾夷扇贝Fosmid克隆混池策略及解码率研究[J]. 中国海洋大学学报:自然科学版, 2019, 49: 44-51. |
| [5] |
华金玲, 施其成, 郁冯艳, 等. 黄淮白山羊瘤胃微生物Fosmid文库的构建与分析[J]. 东北农业大学学报, 2019, 50: 52-58. |
| [6] |
李昂, 吴志明, 周志林, 等. 甘薯近缘种Ipomoea trifida(Kunth)G. Don基因组Fosmid文库构建及PCR筛选体系建立[J]. 华北农学报, 2014, 29: 45-50. |
| [7] |
李昂, 张安, 唐君, 等. Fosmid基因组文库构建及应用现状[J]. 江苏农业科学, 2013, 41: 28-30. |
| [8] |
李碧凤, 朱雅新, 苟潇, 等. 云南大额牛瘤胃宏基因组Fosmid文库的构建与分析[J]. 中国畜牧兽医, 2013, 40: 61-65. |
| [9] |
苏俊, 律娜, 朱宝利, 等. 版纳微型猪近交系清瘦亚系和肥胖亚系猪Fosmid基因组文库的构建[J]. 农业生物技术学报, 2017, 25: 2052-2057. |
| [10] |
吴晓军, 徐丽, 王守才, 等. 玉米基因组Fosmid文库构建及类受体激酶基因(Psy1和Psy2)筛选[J]. 农业生物技术学报, 2014, 22: 1306-1313. DOI:10.3969/j.issn.1674-7968.2014.10.014 |
| [11] |
薛鹏飞, 柳鹏福, 史吉平, 等. 盐单胞菌Fosmid文库构建及群体感应淬灭酶的筛选[J]. 江苏农业科学, 2016, 44: 88-91. |
| [12] |
Liu C, Liu X, Lei L, et al. Fosmid library construction and screening for the maize mutant gene Vestigial glume 1[J]. The Crop Journal, 2016, 4: 55-60. DOI:10.1016/j.cj.2015.09.003 |
| [13] |
Preston CM, Wu KY, Molinski TF, et al. A psychrophilic crenarchaeon inhabits a marine sponge:Cenarchaeum symbiosum gen. nov., sp. nov[J]. PNAS, 1996, 96: 6241-6246. |
| [14] |
Fitz-Gibbon S, Choi AJ, Miller JH, et al. A fosmid-based genomic map and identification of 474 genes of the hyperthermophilic archaeon Pyrobaculum aerophilum[J]. Extremophiles, 1997, 1: 36-51. DOI:10.1007/s007920050013 |
| [15] |
Deckert G, Warren PV, Gaasterland T, et al. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus[J]. Nature, 1998, 392: 353-358. DOI:10.1038/32831 |
| [16] |
Cui Y, Brugiere N, Jackman L, et al. Structural and transcriptional comparative analysis of the S locus regions in two self-incompatible Brassica napus lines[J]. Plant Cell, 1999, 11: 2217-2231. DOI:10.1105/tpc.11.11.2217 |
| [17] |
Bovee D, Zhou Y, Haugen E, et al. Closing gaps in the human genome with fosmid resources generated from multiple individuals[J]. Nature Genetics, 2008, 40: 96-101. DOI:10.1038/ng.2007.34 |
| [18] |
Du H, Yu Y, Ma Y, et al. Sequencing and de novo assembly of a near complete indica rice genome[J]. Nature Communications, 2017, 8: 15324. DOI:10.1038/ncomms15324 |
| [19] |
Zhao Y, Huang J, Wang Z, et al. Allelic diversity in an NLR gene BPH9 enables rice to combat planthopper variation[J]. PNAS, 2016, 113: 12850-12855. DOI:10.1073/pnas.1614862113 |
| [20] |
Zhang CZ, Huang L, Zhang HF, et al. An ancestral NB-LRR with duplicated 3' UTRs confers stripe rust resistance in wheat and barley[J]. Nature Communication, 2019, 10: 4023. DOI:10.1038/s41467-019-11872-9 |
| [21] |
Zimin A, Stevens KA, Crepeau MW, et al. Sequencing and assembly of the 22-gb loblolly pine genome[J]. Genetics, 2014, 196: 875-890. DOI:10.1534/genetics.113.159715 |
| [22] |
Dohm JC, Minoche AE, Holtgrawe D, et al. The genome of the recently domesticated crop plant sugar beet(Beta vulgaris)[J]. Nature, 2014, 505: 546-549. DOI:10.1038/nature12817 |
| [23] |
Suk EK, Schulz S, Mentrup B, et al. A fosmid pool-based next generation sequencing approach to haplotype-resolve whole genomes[M]. New York: Springer New York, 2017.
|
| [24] |
Zou C, Li L, Miki D, et al. The genome of broomcorn millet[J]. Nature Communications, 2019, 10: 436. DOI:10.1038/s41467-019-08409-5 |
| [25] |
Ufarté L, Bozonnet S, Laville E, et al. Functional metagenomics:construction and high-throughput screening of fosmid libraries for discovery of novel carbohydrate-active enzymes[M]. New York: Springer New York, 2016.
|
| [26] |
Riesenfeld CS, Schloss PD, Handelsman J. Metagenomics:genomic analysis of microbial communities[J]. Annual Review of Genetics, 2004, 38: 525-552. DOI:10.1146/annurev.genet.38.072902.091216 |
| [27] |
Kim UJ, Shizuya H, de Jong PJ, et al. Stable propagation of cosmid sized human DNA inserts in an F factor based vector[J]. Nucleic Acids Research, 1992, 20: 1083-1085. DOI:10.1093/nar/20.5.1083 |
| [28] |
萨姆布鲁克, 拉塞尔, 黄培堂, 等. 分子克隆实验指南[M]. .
|
| [29] |
Heavens D, Accinelli GG, Clavijo B, et al. A method to simultaneously construct up to 12 differently sized Illumina Nextera long mate pair libraries with reduced DNA input, time, and cost[J]. Biotechniques, 2015, 59: 42-45. |
| [30] |
Williams LJ, Tabbaa DG, Li N, et al. Paired-end sequencing of Fosmid libraries by Illumina[J]. Genome Research, 2012, 22: 2241-2249. DOI:10.1101/gr.138925.112 |
| [31] |
Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA[J]. Nucleic Acids Research, 1980, 8: 4321-4325. DOI:10.1093/nar/8.19.4321 |