




2. 浙江孚诺医药股份有限公司,东阳 322100;
3. 上海中医药大学中药研究所,上海 201203
2. Zhejiang Fonow Medicine Co. Ltd, Dongyang 322100;
3. Institute of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine, Shanghai 201203
Nur77转录因子,又称为神经生长因子诱导的基因B(Nerve growth factor-induced gene B,NGFI-B)或TR3,是一种类固醇/甲状腺/类视黄醇受体超家族的孤儿核受体蛋白(Orphan nuclear receptor),在多种细胞的信号转导、细胞周期、细胞凋亡的调节中起重要作用[1-3]。作为早期转录因子之一,Nur77定位于细胞核内,起到激活转录,促进细胞生长的作用。因此,Nur77在多种肿瘤细胞中的表达量远超于正常细胞[4]。当受到凋亡信号刺激时,Nur77在磷酸激酶作用下被磷酸化,随之磷酸化的Nur77离开细胞核到达线粒体,与B细胞淋巴瘤-2蛋白(Bcl-2)结合并诱导其构象改变,引发细胞色素C(Cytochrome C)释放,激活半胱氨酸天冬氨酸蛋白酶9(Caspase 9)并进一步激活Caspase级联反应下游的Caspase 3蛋白,最终诱导细胞凋亡[5-6]。由于磷酸化的Nur77直接调控线粒体途径的凋亡过程,因此,围绕以Nur77为作用靶点来进行相关肿瘤治疗药物的开发引起了人们的日益关注[3]。
雷公藤红素(Celastrol)是从传统中药材雷公藤植株中提取的一种药用化合物[7],结构如图 1-A所示,其具有抗氧化、抗癌、抗类风湿,减肥等广泛的生物学活性,尤其是其抗肿瘤活性受到人们的广泛关注[8]。已有研究表明,Celastrol可以抑制肝癌、骨髓癌、乳腺癌、胰腺癌和胃癌等多种人类肿瘤细胞增殖并激活内源性细胞凋亡途径[7, 9-11],通过Bcl-2家族蛋白介导引起线粒体膜通透性改变,释放细胞色素C等凋亡相关因子引发细胞凋亡[12-14]。对诱发细胞凋亡机理的深入研究发现,Celastrol进入细胞后与Nur77结合,促使Nur77从细胞核转移至线粒体,这种易位使NF-κB激酶抑制剂表达增加导致具有抗凋亡活性作用的NF-κB信号因子数量降低,最终诱发细胞凋亡[15-16](图 1-B)。

|
| 图 1 Celastrol结构示意图(A)和Celastrol、Apoptin诱导细胞凋亡的分子机制示意图(B) |
来源于鸡贫血病毒的凋亡蛋白(Apoptin)是一种为数不多的能特异性诱导肿瘤细胞凋亡而不影响正常细胞生长的蛋白。Apoptin进入肿瘤细胞后,能滞留在肿瘤细胞核内,诱发肿瘤细胞凋亡,但其在正常细胞中则会被转运到胞质中降解,因此Apoptin可以诱导众多肿瘤细胞系凋亡而对正常细胞不产生毒副作用[17-18]。类似于Celastrol,Apoptin也是通过促进核内Nur77的磷酸化,使得Nur77从细胞核转移到达线粒体,从而激活内源性凋亡通路,诱导肿瘤细胞凋亡[6, 19](图 1-B)。
由于Celastrol与Apoptin抑制肿瘤生长的作用机理都与Nur77相关,本研究尝试探讨这两种药物联用是否可以通过协同增强Nur77途径的细胞凋亡诱导作用,来加强药物的抗肿瘤药理作用效果。野生型的Apoptin由于其本身多肽链一级结构的原因,在通过原核表达系统重组表达时大都以包涵体形式表达,易产生聚集沉淀现象[5, 20-21]。本课题组在前期研究中制备了一种Apoptin的突变体tApoptin,实现了在原核表达系统中的可溶性表达,并且在商陆皂苷甲(EsA)存在下表现出显著的抗肿瘤效果[22](简称为tApoptinE)。本研究首先通过MTT实验考察了Celastrol与tApoptinE两者联用是否对不同肿瘤细胞的增殖抑制存在协同作用,随后通过流式凋亡分析和免疫印迹(Western blot)技术进一步分析了Celastrol与tApoptinE联用后抗肿瘤活性的分子机制。
1 材料与方法 1.1 材料人肝癌细胞(SMMC-7721,简称SMCC)、人宫颈癌细胞(HeLa)、人非小细胞肺癌细胞(A549)、人胚肺细胞(MRC-5)、tApoptin-pET28a均为本实验保存。表达宿主菌为E.coli BL21(DE3)。卡那霉素(Kan)、异丙基-β-D-硫代吡喃半乳糖苷(IPTG)、噻唑蓝(MTT)等均购自碧云天公司。商陆皂苷甲(EsA)购自新铂化学技术有限公司。
1.2 方法 1.2.1 体外细胞培养SMMC、HeLa、A549和MRC-5细胞均采用RPMI-1640培养基(10%胎牛血清,1%青霉素-链霉素)。所有细胞置于含5% CO2,37℃培养箱中培养。
1.2.2 药物体外生长抑制实验(MTT法)各种细胞按1×104/孔接种至96孔细胞板中,培养24 h。检测单药对SMMC细胞生长抑制作用时,将不同浓度(62.5、125、250、500、1 000 nmol/L)的Celastrol或tApoptinE分别与SMMC细胞孵育24 h,tApoptinE为混合了50 μmol/L EsA的tApoptin;检测联合用药作用效果时,分别在不同浓度的Celastrol(75、150、300、600、1 200 nmol/L)存在下,浓度梯度的tApoptinE(62.5、125、250、500、100 nmol/L)与SMMC细胞孵育24 h,Celastrol与tApoptinE混合后同时与细胞共孵育。然后弃掉细胞板中培养基,每孔加入200 μL含5 mg/mL MTT的溶液后培养箱中继续培养2 h。弃掉MTT溶液,每孔加入150 μL二甲基亚砜(DMSO),振动溶解甲瓒,以Thermo酶标仪(吸收波长为490 nm)测定吸光度,计算细胞存活率。
1.2.3 流式细胞术凋亡分析每孔1×104个SMMC细胞接种到24孔板中,于含5% CO2,37℃培养箱中培养24 h。对照组中分别加入600 nmol/L Celastrol,65、125、250 nmol/L tApoptinE;实验组分别为与600 nmol/L Celastrol混合的65、125、250 nmol/L tApoptinE。药物与细胞共孵育24 h之后收获各组细胞并进行两次PBS洗涤,以100 μL结合缓冲液重悬细胞,加入10 μL Annexin V-FITC和碘化丙啶(PI)1:1混合液,20℃黑暗孵育细胞20 min,加入400 μL结合缓冲液。通过FACScan流式细胞仪对经处理的样品进行分析。
1.2.4 免疫印迹分析每皿1×107个SMMC细胞接种到55 cm2的培养皿中,于含5% CO2,37℃培养箱中培养12 h至贴壁。每组分别加入600 nmol/L Celastrol,65、125、250 nmol/L tApoptinE和62.5 nmol/L tApoptinE + 600 nmol/L Celastrol、125 nmol/L tApoptinE + 600 nmol/L Celastrol、250 nmol/L tApoptinE + 600 nmol/L Celastrol,并设置一组无试剂添加的空白对照组。药物与细胞共孵育24 h后收获细胞,裂解细胞获得全细胞裂解物,12 000 r/min离心15 min取上清蛋白。通过BCA蛋白定量试剂盒测定各组上清蛋白浓度,然后再用裂解液将各组上清稀释至同一蛋白浓度。以SDS上样缓冲液处理稀释后细胞裂解液,之后进行SDS聚丙酰胺凝胶蛋白电泳分离样品,然后将凝胶泳道中的蛋白转移至聚偏氟乙烯(PVDF)膜。之后,将膜置于含5%脱脂奶粉的封闭液中常温封闭过夜,再以PBS洗涤膜3次。将封闭处理后的膜与特异性一抗4℃孵育8 h,以PBS洗涤膜3次。随后将与一抗孵育后的膜与辣根过氧化物酶缀合的二抗室温孵育1 h,以PBS洗涤3次。最后用ECL化学发光超敏显色试剂盒对膜进行显像,并用化学成像系统拍照。
1.2.5 联合作用指数(Combined index,CI)计算两种药物联用是否具有协同效应是通过CI值来判断的,CI值小于1则说明药物联用具有协同作用[23]。在Compusyn软件(Version 1.0,Combo SynInc,USA)中分别输入单独用药和联合用药各组药物的浓度和细胞抑制率来计算CI值来判断药物间协同作用的大小。
2 结果 2.1 联合用药对肿瘤细胞生长的抑制作用的MTT法分析为了分析Celastrol与tApoptinE两者联用是否能对肿瘤细胞的生长产生协同抑制作用,固定浓度的Celastrol与tApoptinE联用,利用细胞生长抑制实验检测二者联合的作用效果。分别在不同Celastrol的固定浓度下(0、75、150、300、600、1 200 nmol/L)观察tApoptinE浓度变化(62.5-1 000 nmol/L)对肿瘤细胞SMMC生长抑制效应,MTT结果显示,Celastrol提高了tApotpinE对SMMC细胞的生长抑制活性,并且该效果具有浓度依赖性:低浓度的Celastrol(75、150 nmol/L)对tApoptinE的作用效果基本没有影响;当Celastrol达到300 nmol/L时一定程度上提高了tApoptinE的抗肿瘤活性,tApoptinE对SMMC细胞的生长抑制能力从Celastrol不存在时的IC50值717.7 nmol/L下降到270.1 nmol/L;当Celastrol ≥ 600 nmol/L时(600、1 200 nmol/L)显著提升了tApoptinE对SMMC细胞的生长抑制作用,tApoptinE对SMMC细胞的IC50值分别降至48.63、45.74 nmol/L(图 2-A,表 1)。这些结果表明,≥ 300 nmol/L的Celastrol与不同浓度tApoptinE联合用药可对SMMC细胞的生长活性产生更好的抑制效果。

|
| 以上数据来源于3次实验(n=3),下同 图 2 Celastrol与tApoptinE单独及联合用药24 h对SMMC细胞的生长抑制效果分析(A)及联合用药协同指数CI值(B) |
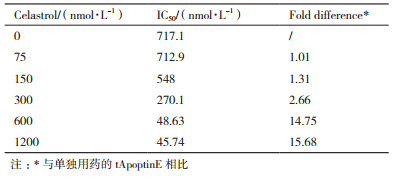
CI值可以判断两种药物联用是否具有协同效应,CI值小于1则说明药物联用具有协同作用。CI值计算结果显示,低浓度Celastrol(75、150 nmol/L)与不同浓度tApoptinE联合用药其CI值1左右,尚未表现出协同作用;当Celastrol达到300 nmol/L时,CI值明显小于1,体现出了一定的协同效应;当Celastrol为600 nmol/L时,CI值最低(图 2-B)。这说明当tApoptinE与高于300 nmol/L的Celastrol联用时,这两种药物的联用存在显著的协同效应且Celastrol为600 nmol/L时协同效果最佳。因此后续实验选取CI值最低的600 nmol/L Celastrol作为联用的固定浓度。
2.2 联合用药抑制肿瘤细胞生长的流式细胞术分析为了阐明Celastrol与tApoptinE联用后对肿瘤细胞的协同抑制效应的分子机制,我们以Annexin V-FITC/FACS方法对联合用药后的肿瘤细胞凋亡率进行了检测分析。如图 3所示,左上的Q1区代表含有独立细胞核但是细胞膜破损严重的细胞,左下的Q4区代表正常的活细胞,右上的Q2区代表晚期凋亡或者死亡的细胞,右下的Q3区代表早期凋亡的细胞,凋亡率为早期凋亡率(Q3)与晚期凋亡率(Q2)结果相加,经计算后得到如下结果:与SMMC细胞孵育24 h后,单独使用Celastrol(600 nmol/L)处理的,仅产生29.2%的细胞凋亡率;单独使用62.5 nmol/L、125 nmol/L和250 nmol/L浓度的tApoptinE时,诱导SMMC的细胞凋亡率分别为22.09%、33.61%和43.9%;而当上述相应浓度tApoptinE与600 nmol/L Celastrol联合用药时,诱导细胞的凋亡率分别达到了71.6%、79.2%和76.7%(图 3-A-B),即在不同tApoptinE浓度下的诱导凋亡率均提高了2-3倍。因此,Celastrol与tApoptinE联用后的生长抑制作用的增强是通过提升对肿瘤细胞的诱导凋亡效应来实现的。

|
| 图 3 联合用药对SMMC细胞的流式细胞术凋亡检测(A)及SMMC细胞的凋亡率直方图(B) |
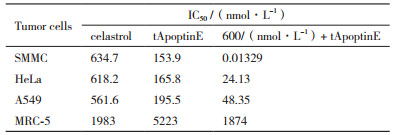
上述流式细胞术凋亡分析实验的结果表明Celastrol与tApoptinE联合用药在SMMC细胞中存在协同作用的原因使Celastrol与tApoptinE联用后强化了对SMMC细胞的凋亡诱导效应。因为这两种药物都是通过Nur77的磷酸化来激活细胞凋亡通路,所以本课题组进一步通过免疫印迹实验来比较分析这种诱导作用的增强是否与Nur77的表达量相关。分析MRC-5、SMMC、HeLa和A549四种细胞的Nur77表达量,发现Nur77在SMMC、HeLa和A549三种肿瘤细胞中的表达量显著超过在正常细胞MRC-5中的表达量(图 4-B);而这种差异性与MTT实验结果显示的两种药物联合用药对细胞生长的抑制作用效果呈正相关:在相同用药浓度下,对正常细胞的生长几乎无生长抑制作用,但对所有测试的肿瘤细胞(SMMC、HeLa和A549细胞)均有明显的协同抑制作用(图 4-A),与600 nmol/L celastrol联用后tApoptinE对SMMC、HeLa和A549细胞的IC50值分别为0.013 29、24.13、48.35 nmol/L,在SMMC细胞中表现最高抑制活性。因此,Celastrol与tApoptinE无论是单独用药还是联合用药的协同抑制作用效果与Nur77表达量直接相关。

|
| 图 4 单独或联合用药对不同细胞生长的抑制作用其中对照组为无任何药物处理,只含有培养基的细胞(A)及不同细胞内的Nur77表达量(B) |
上述实验证明了Celastrol与tApoptinE的联合用药导致的细胞生长抑制效应与胞内Nur77的表达量直接相关,本研究进一步通过免疫印迹实验检测了胞内磷酸化的Nur77(p-Nur77)的蛋白水平。结果如图 5所示,与空白对照组相比,单独使用Celastrol或tApoptinE处理后,可显著上调细胞内p-Nur77的表达;而当Celastrol与tApoptinE联用后,又进一步上调了p-Nur77的表达。这一结果表明Celastrol与tApoptinE联用后进一步诱导Nur77发生磷酸化,促使NuR77传递凋亡信号,从而实现加强诱导细胞凋亡效果。

|
| 图 5 单独及联合用药后SMMC细胞中Nur77表达量及磷酸化水平(A)及蛋白量的直方图(B) |
为了明确磷酸化的Nur77诱导细胞凋亡更具体的分子机制,我们分析了联合用药后在细胞线粒体凋亡过程中起重要作用的Caspase家族蛋白和Bcl-2家族蛋白的表达量。
若Caspase 3、Caspase 8和Caspase 9表达水平下调则预示着级联反应被激活、细胞凋亡启动。Western blot实验结果显示,在经单独的Celastrol或tApoptinE处理后这3种蛋白的表达水平相对于空白对照组均有所下调,并且在Celastrol与tApoptinE联合处理的细胞中,3种蛋白表达量的下调更加显著(图 6-A,6-C)。同时,Bax/Bcl-2比值上升时可以激活下游线粒体死亡级联反应、引发细胞凋亡,在经过单独的Celastrol或tApoptinE处理后Bax/Bcl-2比值增大,并且,在联合用药组中该现象更加明显(图 6-B,6-D)。上述结果表明,Celastrol与tApoptinE单独用药时可以调控Caspase和Bcl-2家族蛋白的表达,激活细胞的线粒体凋亡通路,而两者联用后产生协同作用,使得该通路被进一步激活。并且,上述过程伴随Nur77磷酸化进行,暗示磷酸化的Nur77通过调控Caspase和Bcl-2家族蛋白的表达诱导细胞凋亡。

|
| 图 6 单独及联合用药后SMMC细胞中Caspase家族蛋白表达量(A)和蛋白量直方图(C)及Bcl-2家族蛋白表达量(B)和蛋白量直方图(D) |
结合两种或者两种以上治疗药物的联合用药法因具有临床优势而成为肿瘤治疗的常规方式[24],除了降低新药研发成本,联合用药还可以克服很多单药治疗带来的局限性[25]。首先,联合用药的药物会以特征性协同或者相加的方式作用于多种关键途径,由此取得更好的抗肿瘤效果[26];其次,联合用药可以解决由于单一化合物持续治疗,从而诱导癌细胞招募替代的挽救途径引起的耐药性问题,增加药物在胞内积累[27];疗效的提升和更少耐药性的诱导效应使得单一药物的使用剂量总体都得以降低,从而减轻了药物所带来的全身性毒性[28];最后,联合用药的应用降低了相同疗效下单药治疗的不良反应。因此,探索肿瘤治疗的新的联合用药方式具有非常重要的实际应用价值。对于Celastrol来说,He等[29]曾发现Celastrol可以抑制藤黄酸(Gambogic acid,GA)在发挥抗肿瘤作用中引发的NF-κB激活,从而增强肿瘤细胞对GA的敏感性,取到增强疗效的效果。
作为一种已研究比较全面的五环三萜类化合物,Celastrol是中药雷公藤(Thunder God Vine)中的一种重要生物活性成分[30]。在过去的10年里,越来越多的研究强调Celastrol在不同临床领域的医用价值[31]。数据表明,由于能够抑制NF-κB途径,干扰促炎细胞因子和趋化因子的产生,以及炎症细胞的迁移、增殖和活化,Celastrol作为抗炎化合物治疗风湿性关节炎、系统性红斑狼疮、骨关节炎和过敏等疾病具有巨大潜力[32-35];Celastrol可以通过抑制NF-κB和HSP来治疗神经退行性疾病(例如阿尔兹海默症)[36];最新数据显示Celastrol还可以应用在糖尿病、肥胖症、动脉粥样硬化和听力丧失等疾病的治疗中[37-40]。此外,Celastrol的抗癌特性一直备受关注,Celastrol可以应用于肝癌、骨髓癌、乳腺癌、胰腺癌和胃癌等多种肿瘤的治疗,这主要是由于其能够抑制转录因子(如NF-κB和HIF-1α),以及蛋白质稳态的介质(如HSP90伴侣)[41-42]。但是,当应用到肿瘤临床治疗时,Celastrol的使用仍受到限制:一方面,Celastrol的长期使用伴随着全身毒性和多种不良反应,如体重减轻、血液毒性、肝损伤和生殖障碍[43];另一方面,Celastrol在细胞内发挥作用时所产生的其他反应不是我们期望的,例如,Zheng等[44]发现Celastrol会下调维持细胞黏附和抑制细胞迁移功能的钙黏蛋白(E-cadherin)的表达,从而降低肿瘤细胞对Celastrol的敏感性。因此,我们如果能通过药物联用的方式在保证发挥Celastrol抗肿瘤治疗效果的同时,降低Celastrol的用量,就可能减少Celastrol的临床应用所带来的毒性反应和其他不良反应。
由鸡贫血病毒的VP3基因编码的Apoptin具有在不损害正常细胞的基础上特异性杀死肿瘤细胞的特性[18]。但是,尽管Apoptin的这种选择性抗肿瘤的能力在上世纪末就被发现,但20多年来由于Apoptin本身的一级结构特点(N端富集亮氨酸,蛋白易产生聚集沉淀),给它的表达纯化带来困难,限制了其应用[21]。有研究表明,去掉Apoptin N端的43个氨基酸后获得的突变体tApoptin可以在原核细胞中实现可溶性表达,并且仍对肿瘤细胞保持生长抑制活性,但引起细胞凋亡的效应有所降低[20]。如果要保持tApoptin原有的抗肿瘤活性,就需要大大提高用药量。因此,我们课题组前期通过tApoptin与EsA联合用药的方式(tApoptinE),使得tApoptin的药效显著提高[22]。
由于Celastrol与tApoptin都以Nur77途径诱导细胞凋亡起到抗肿瘤作用,所以我们将这两种药物联用。MTT结果显示,浓度固定的Celastrol与tApoptinE联用后可以提升tApoptin的抗肿瘤效果,尤其是同较低浓度的Celastrol(75、150、300 nmol/L)联用相比较,tApoptinE与600 nmol/L或者1 200 nmol/L的Celastrol联合用药更加显著地提高单独tApoptinE对肿瘤细胞SMMC的生长抑制作用,并且这两组的药效提升效果接近。这可能是由于Celastrol本身对细胞抑制活性较弱,所以当低浓度的Celastrol与tApoptinE联用时由于Celastrol浓度较低不能很好地发挥提高tApoptinE活性的作用,而当Celastrol提高到一定浓度时,二者联合对细胞生长有较强的抑制作用,药效达到峰值很难再提升。
CI值小于1表示两药具有协同作用[45],当与tApoptinE联用的Celastrol浓度高于300 nmol/L时,联用组CI值均小于1,这说明在某些浓度范围内Celastrol与tApoptinE联用后确实存在协同作用。此外,600 nmol/L Celastrol与tApoptinE联用后,CI计算所得的值为所有Celastrol组中最低,CI值越低表示协同效果越好[46],因此可以判断,600 nmol/L Celastrol与tApoptinE联用后协同作用在所有联用组中最强。文献报道在一些特定的情况下,两种药物的协同效应仅在一定的药物浓度范围内才能体现,特别是其中一种药物浓度过高易使得协同效果反而不明显[47],据此推断,当Celastrol浓度过低时(75、150 nmol/L),Celastrol尚未达到与tApoptinE协同抗肿瘤的条件,因此两药联用不表现正协同作用;当Celastrol浓度高达1 200 nmol/L时,单药对肿瘤细胞的生长抑制作用已非常强,所以协同效果不如600 nmol/L Celastol。考虑到Celastrol的细胞毒性[43],在保障协同作用及药效的情况下尽可能地降低Celastrol用量,因此我们选择600 nmol/L的Celastrol作为固定联用浓度。
对联合用药的流式凋亡分析实验表明,协同作用加强了对细胞的凋亡诱导效应。通过Western blot实验分析MRC-5、SMMC、HeLa和A549四种细胞的Nur77表达量,并结合Celastrol(600 nmol/L)与不同浓度tApoptinE联用对上述四种细胞的MTT实验结果显示,在相同用药浓度下,联合用药对Nur77低表达的正常细胞不表现协同抑制作用,生长抑制作用小;但对Nur77高表达的肿瘤细胞(SMMC、HeLa和A549细胞)均有明显的协同抑制作用,生长抑制作用强。进一步表明这两种药物的细胞生长抑制作用必须通过Nur77这个靶点才能实现。此外,Celastrol与tApoptinE联用具有一定的靶向作用效果,其对Nur77高表达的细胞更具杀伤性。这种增强诱导细胞凋亡效应直接与Nur77的表达量呈正相关。
Nur77途径的诱导凋亡效应只有在Nur77磷酸化,并且磷酸化的Nur77移动到线粒体才能进行触发内源性细胞凋亡[48]。从Western blot的结果可以看到Celastrol或tApoptinE单独给药后均可以诱导Nur77发生磷酸化,上调Bax/Bcl-2比值,活化Caspase 9以及下游的Caspase 3,引发细胞凋亡[49]。这一方面证实了Celastrol与tApoptinE在胞内发挥诱导细胞凋亡作用是通过共同作用于Nur77通路来实现的;另一方面也显示了联合用药组Nur77的磷酸化水平提高更为显著,引起Bcl-2家族蛋白和Caspase家族蛋白相关变化更加显著。因此Celastrol与tApoptinE的联合用药可利用两种药物间的正协同效应提升抗肿瘤作用效果,降低这两种药物各自的使用量,从而有可能减少不良反应的发生,促进这两种药物的临床应用开发进程。
4 结论本研究证实Celastrol与tApoptinE联合用药对多种Nur77高表达的肿瘤细胞存在协同抑制作用,能够在提升药效的同时降低用药剂量,联用的药物协同作用于同一条Nur77诱发的细胞凋亡通路,强化激活线粒体凋亡途径,诱导细胞凋亡。本研究提出的联用方案为联合用药治疗癌症提供了新的策略。
| [1] |
Wu H, Li XM, Wang JR, et al. NUR77 exerts a protective effect against inflammatory bowel disease by negatively regulating the TRAF6/TLR-IL-1R signalling axis[J]. J Pathol, 2016, 238(3): 457-469. |
| [2] |
Cortez-Toledo O, Schnair C, Sangngern P, et al. Nur77 deletion impairs muscle growth during developmental myogenesis and muscle regeneration in mice[J]. PLoS One, 2017, 12(2): 1-17. |
| [3] |
Liu TY, Yang XY, Zheng LT, et al. Activation of Nur77 in microglia attenuates proinflammatory mediators production and protects dopaminergic neurons from inflammation-induced cell death[J]. Neurochem, 2017, 140(4): 589-604. DOI:10.1111/jnc.13907 |
| [4] |
Wu L, Chen L. Characteristics of Nur77 and its ligands as potential anticancer compounds(Review)[J]. Mol Med Rep, 2018, 18(6): 4793-4801. |
| [5] |
Hou Z, Mao J, Lu Y, et al. rApoptin induces apoptosis in human breast cancer cells via phosphorylation of Nur77 and Akt[J]. Biochem Biophys Res Commun, 2018, 498(1): 221-227. DOI:10.1016/j.bbrc.2018.02.204 |
| [6] |
Maddika S. Cancer-specific toxicity of apoptin is independent of death receptors but involves the loss of mitochondrial membrane potential and the release of mitochondrial cell-death mediators by a Nur77-dependent pathway[J]. Cell Sci, 2005, 118(19): 4485-4493. DOI:10.1242/jcs.02580 |
| [7] |
Mou H, Zheng Y, Zhao P, et al. Celastrol induces apoptosis in non-small-cell lung cancer A549 cells through activation of mitochondria- and Fas/FasL-mediated pathways[J]. Toxicol Vitr, 2011, 25(5): 1027-1032. DOI:10.1016/j.tiv.2011.03.023 |
| [8] |
Li X, Wang H, et al. Celastrol strongly inhibits proliferation, migration and cancer stem cell properties through suppression of Pin1 in ovarian cancer cells[J]. Eur J Pharmacol, 2019, 842: 146-156. DOI:10.1016/j.ejphar.2018.10.043 |
| [9] |
Wang Q, Yu X, Li F, et al. Efficacy of celastrol combined with cisplatin in enhancing the apoptosis of U-2OS osteosarcoma cells via the mitochondrial and endoplasmic reticulum pathways of apoptosis[J]. Oncol Lett, 2019, 17(3): 3305-3313. |
| [10] |
Lin FZ, Wang SC, Hsi YT, et al. Celastrol induces vincristine multidrug resistance oral cancer cell apoptosis by targeting JNK1/2 signaling pathway[J]. Phytomedicine, 2019, 54: 1-8. DOI:10.1016/j.phymed.2018.09.181 |
| [11] |
Yu X, Zhou X, Fu C, et al. Celastrol induces apoptosis of human osteosarcoma cells via the mitochondrial apoptotic pathway[J]. Oncol Rep, 2015, 34(3): 1129-1136. |
| [12] |
Greil R, Anether G, Johrer K, et al. Tracking death dealing by Fas and TRAIL in lymphatic neoplastic disorders: pathways, targets, and therapeutic tools[J]. Leukoc Biol, 2003, 74(3): 311-330. |
| [13] |
Los M, Wesselborg S, Schulze-Osthoff K. The role of caspases in development, immunity, and apoptotic signal transduction: lessons form knockout mice[J]. Immunity, 1999, 10(6): 629-639. DOI:10.1016/S1074-7613(00)80062-X |
| [14] |
Tait SWG, Green DR. Mitochondria and cell death: Outer membrane permeabilization and beyond[J]. Nat Rev Mol Cell Biol, 2010, 11(9): 621-632. DOI:10.1038/nrm2952 |
| [15] |
Hu M, Luo Q, Alitongbieke G, et al. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy[J]. Mol Cell, 2017, 66(1): 141-153. |
| [16] |
Pei L, Castrillo A, Tontonoz P. Regulation of cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3 hui by the orphan nuclear receptor Nur77[J]. Mol Endocrinol, 2005, 20(4): 786-794. |
| [17] |
Burek M, Schulze-Osthoff K, Los M, et al. Apoptin-induced cell death is modulated by Bcl-2 family members and is Apaf-1 dependent[J]. Oncogene, 2005, 25(15): 2213-2222. |
| [18] |
Castro J, Ribo M, Benito A, et al. Apoptin, A versatile protein with selective antitumor activity[J]. Curr Med Chem, 2018, 25(30): 3540-3559. DOI:10.2174/0929867325666180309112023 |
| [19] |
Chaabane W, Cieślar-Pobuda A, El-Gazzah M, et al. Human-gyrovirus-apoptin triggers mitochondrial death pathway-Nur77 is required for apoptosis triggering[J]. Neoplasia, 2014, 16(9): 679-693. DOI:10.1016/j.neo.2014.08.001 |
| [20] |
Shen Ni L, Allaudin ZN bt, et al. Selective apoptosis induction in MCF-7 cell line by truncated minimal functional region of Apoptin[J]. BMC Cancer, 2013, 13: 488-497. DOI:10.1186/1471-2407-13-488 |
| [21] |
Vilanova M, Castro J, Benito A, et al. A truncated apoptin protein variant selectively kills cancer cells[J]. Invest New Drugs, 2017, 35(3): 260-268. DOI:10.1007/s10637-017-0431-6 |
| [22] |
李柳美, 曹雪玮, 王晓旦, 等. 商路皂苷甲(ESA)的联用可显著提高tApoptin凋亡蛋白的抗肿瘤活性[J]. 中国生物化学与分子生物学报, 2019, 35(10): 1098-1107. |
| [23] |
Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies[J]. Pharmacol Rev, 2006, 58: 621-681. DOI:10.1124/pr.58.3.10 |
| [24] |
DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs[J]. Health Econ, 2003, 22: 151-85. DOI:10.1016/S0167-6296(02)00126-1 |
| [25] |
Bozic I, Reiter JG, et al. Evolutionary dynamics of cancer in response to targeted combination therapy[J]. eLife, 2013, 2: 1-15. |
| [26] |
Mokhtari RB, Homayouni TS, Baluch N, et al. Combination therapy in combating cancer[J]. Oncotarget, 2017, 8(23): 22-43. |
| [27] |
Khdair A, Chen D, Patil Y, et al. Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance[J]. Control Release, 2010, 141: 137-144. DOI:10.1016/j.jconrel.2009.09.004 |
| [28] |
DeVita VT Jr, Young RC, Canellos GP. Combination versus single agent chemotherapy: a review of the basis for selection of drug treatment of cancer[J]. Cancer, 1975, 35: 98-110. DOI:10.1002/1097-0142(197501)35:1<98::AID-CNCR2820350115>3.0.CO;2-B |
| [29] |
He D, Xu Q, Yan M, et al. The NF-kappa B inhibitor, celastrol, could enhance the anti-cancer effect of gambogic acid on oral squamous cell carcinoma[J]. BMC Cancer, 2009, 9: 343-351. DOI:10.1186/1471-2407-9-343 |
| [30] |
Chin K, Pang K, et al. Anti-inflammatory nutraceuticals and chronic diseases[J]. Adv Exp Med Biol, 2016, 928: 97-130. |
| [31] |
Cascão R, Fonseca JE, Moita LF. Celastrol: A spectrum of treatment opportunities in chronic diseases[J]. Frontiers in Medicine, 2017, 4: 69-86. DOI:10.3389/fmed.2017.00069 |
| [32] |
Gan K, Xu L, Feng X, et al. Celastrol attenuates bone erosion in collagen-Induced arthritis mice and inhibits osteoclast differ- entiation and function in RANKL-induced RAW264. 7[J]. Int Immunopharmacol, 2015, 24(2): 239-246. |
| [33] |
Xu X, Wu Z, Xu C, et al. Observation on serum anti-double stranded DNA antibodies of tripterine in systemic lupus erythematosus of(NZBxW)F1 mice[J]. Ann Rheum Dis, 2003, 62(4): 377-378. DOI:10.1136/ard.62.4.377 |
| [34] |
Ding QH, Cheng Y, Chen WP, et al. Celastrol, an inhibitor of heat shock protein 90beta potently suppresses the expression of matrix metalloproteinases, inducible nitric oxide synthase and cyclooxygenase-2 in primary human osteoarthritic chondrocytes[J]. Eur J Pharmacol, 2013, 708: 1-7. DOI:10.1016/j.ejphar.2013.01.057 |
| [35] |
Kim Y, Kim K, Lee H, et al. Celastrol binds to ERK and inhibits FcepsilonRI signaling to exert an anti-allergic effect[J]. Eur J Pharmacol, 2009, 612: 131-142. DOI:10.1016/j.ejphar.2009.03.071 |
| [36] |
Paris D, Ganey NJ, Laporte V, et al. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer's disease[J]. Neuroinflammation, 2010, 7: 17-31. DOI:10.1186/1742-2094-7-17 |
| [37] |
Abu Bakar MH, Cheng KK, Sarmidi MR, et al. Celastrol protects against antimycin A-induced insulin resistance in human skeletal muscle cells[J]. Molecules, 2015, 20(5): 8242-8269. DOI:10.3390/molecules20058242 |
| [38] |
Liu J, Lee J, Salazar Hernandez MA, et al. Treatment of obesity with celastrol[J]. Cell, 2015, 161(5): 999-1011. DOI:10.1016/j.cell.2015.05.011 |
| [39] |
Zhu F, Li C, Jin XP, et al. Celastrol may have an anti-atherosclerosis effect in a rabbit experimental carotid atherosclerosis model[J]. Int J Clin Exp Med, 2014, 7(7): 1684-1691. |
| [40] |
Selimoglu E. Aminoglycoside-induced ototoxicity[J]. Curr Pharm Des, 2007, 13(1): 119-126. DOI:10.2174/138161207779313731 |
| [41] |
Ji N, Li J, Wei Z, et al. Effect of celastrol on growth inhibition of prostate cancer cells through the regulation of hERG channel in vitro[J]. Biomed Res Int, 2015, 15: 475-481. |
| [42] |
Gu L, et al. Celastrol prevents atheroscle- rosis via inhibiting LOX-1 and oxidative stress[J]. PLoS One, 2013, 8(6): e65477. DOI:10.1371/journal.pone.0065477 |
| [43] |
Zhu H, Ding WJ, Wu R, et al. Synergistic anti- cancer activity by the combination of TRAIL/ APO-2L and celastrol[J]. Cancer Invest, 2010, 28: 23-32. DOI:10.3109/07357900903095664 |
| [44] |
Zheng L, Fu Y, Zhuang L, et al. Simultaneous NF-κB inhibition and E-cadherin upregulation mediate mutually synergistic anticancer activity of celastrol and SAHA in vitro and in vivo[J]. Int J Cancer, 2014, 135(7): 1721-1732. DOI:10.1002/ijc.28810 |
| [45] |
Zhang N, Fu JN, Chou TC. Synergistic combination of microtubule targeting anticancer fludelone with cytoprotective panaxytriol derived from panax ginseng against MX-1 cells in vitro: Experimental design and data analysis using the combination index method[J]. American Journal of Cancer Research, 2016, 6: 97-104. |
| [46] |
Soriano AF, Helfrich B, Chan DC, et al. Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines[J]. Cancer Res, 1999, 59(24): 6178-6184. |
| [47] |
Gupta V, Dixit NM. Trade-off between synergy and efficacy in combinations of HIV-1 latency-reversing agents[J]. PLoS Comput Biol, 2018, 14: 1-21. |
| [48] |
Wang A, Rud J, Olson CM, et al. Phosphorylation of Nur77 by the MEK-ERK-RSK cascade induces mitochondrial translocation and Apoptosis in T cells[J]. Immunol, 2009, 183: 3268-3277. DOI:10.4049/jimmunol.0900894 |
| [49] |
Agostini-Dreyer A, Jetzt AE, Stires H, et al. Endogenous IGFBP-3 mediates intrinsic apoptosis through modulation of Nur77 phosphorylation and nuclear export[J]. Endocrinology, 2015, 156: 4141-4151. DOI:10.1210/en.2015-1215 |