




2. 农业部肉羊遗传育种重点实验室,呼和浩特 010018;
3. 内蒙古自治区山羊遗传育种工程技术研究中心,呼和浩特 010018;
4. 内蒙古金莱牧业科技有限责任公司,呼和浩特 010018
2. Key Laboratory of Mutton Sheep Genetics and Breeding, Ministry of Agriculture, Hohhot 010018;
3. Engineering Research Center for Goat Genetics and Breeding, Inner Mongolia Autonomous Region, Hohhot 010018;
4. Inner Mongolia Jinlai Livestock Technology Co., Ltd, Hohhot 010018
基于考古学和遗传学等方法的研究表明,家山羊是约在10 000年前的新石器时代由西亚肥沃新月地带的野山羊(Bezoars,Capra aegagrus)驯化而来,是最早驯化的反刍动物之一[1-2]。随着人类的迁徙与演化,山羊是目前全球范围内分布最广泛的牲畜物种之一,主要用于生产肉、奶、皮和毛(绒)等农业生产资源[3-4]。据统计资料显示,全世界范围内共有10亿多只不同生产用途的山羊饲养在各种生态区内,超过90%的山羊分布在亚洲和非洲;其次是美洲、欧洲和大洋洲,包括肉用、乳用、皮毛用、绒毛用和普通山羊等不同生产用途的576个山羊品种(http://www.fao.org/faostat/en/)[4-5]。山羊是发展中国家农牧民重要的家畜之一,但相对于奶牛、家猪、绵羊和家马等经济效益较高的牲畜品种,山羊的分子生物学研究和遗传育种工作总体相对落后,严重阻碍了发展中国家贫困偏远地区的经济发展[6]。
随着人类基因组计划的实施与完成,单核苷多态性(Single-nucleotide polymorphism,SNP)因具有数量多,分布广泛,易于快速、规模化筛查,便于基因分型等特点,已成为动物种质资源遗传多样性评估和基因功能定位研究的有力工具[7-9]。高通量测序技术的应用极大地促进了家畜基因组组装和遗传变异检测的研究[10-11],如家牛[12]、家马[13]、家猪[14]、山羊[15]和绵羊[16]等参考基因组组装以及第一款家牛商业化芯片的研制[17]。2010年,国际山羊基因组协会(International goat genome consortium,IGGC)成立,标志着高通量测序(Next generation sequencing,NGS)技术开始广泛应用于山羊的基因组研究[18-19];2013年完成了世界上首个山羊参考基因组草图[15],并推出Goat SNP50K磁珠芯片[20]和66K目标捕获芯片[21]。2017年,Bickhart等[22]组装的近乎完整的参考基因组精细图谱ARS1,为山羊功能基因的精细定位提供了更加可靠的基因组信息。通过对基因组的重测序、简化基因组测序、外显子测序和RNA-seq等技术方法,与参考基因组(CHIR_1.0、CHIR_2.0和ARS1)比对,获得了大量的遗传变异信息,为更全面的揭示山羊的遗传多样性、环境适应以及人工选择反应提供了遗传标记信息。因此,本文主要对山羊基因图谱(遗传图谱、物理图谱、转录图谱与表达图谱)以及分子遗传变异信息的检测进展进行了综述,以期为进一步利用参考基因组信息和遗传变异标记对山羊进行经济性状的遗传基础研究和分子育种提供参考。
1 山羊基因组图谱 1.1 遗传图谱1996年,Vaiman等[23]基于微卫星标记和共线性分析,利用12个半同胞家系山羊(萨能奶山羊和阿尔卑斯山羊杂交种)构建得到了低分辨率的连锁图谱,并利用荧光原位杂交技术(Fluorescence in situ hybrid,FISH)确定了204个微卫星标记;最终得到全长为2 300 cM的连锁图谱,覆盖了山羊基因组长度的80%。1998年,Schibler等[24-25]构建了山羊BAC文库,并通过ZOO-FISH技术在山羊染色体上定位了202个基因,同时在已有的山羊遗传图谱上增加了30个微卫星标记,以此构建的细胞遗传-遗传连锁合成图含有307个微卫星标记257个基因,遗传图谱长度约2 737 cM,覆盖了山羊基因组的88%。2005年,Maddox[26]对绵羊和山羊的遗传图谱进行比较,结果显示有218个公共共有基因座,同时发现它们的同源基因座在图中的位置很一致。
1.2 物理图谱1975年,Goss和Harris[27]共同创立了体细胞杂交技术,即辐射杂交(Radiation hybrid,RH)基因组作图技术,其原理是用辐射来诱导染色体断裂,并将辐射过的细胞与正常细胞进行杂交,获得含有染色体片段的杂种细胞。随后利用辐射杂交技术成功在人类、家属、家牛等不同物种中构建了基因组长范围的高分辨连续物理图谱,极大了促进了人类、小鼠及不同家畜物种的基因组研究进展。Du等[28-29]利用辐射杂种嵌板技术,首次构建了山羊全基因组辐射杂种图谱(CHIRH5000),为标记密度最高的的辐射杂种图谱。随后,更多的标记定位在山羊细胞遗传-遗传连锁合成图,这些研究和相应建成的山羊图谱数据(http://locus.jouy.inra.fr)加深对哺乳动物染色体进化的了解,加速反刍动物图位克隆的研究[30]。
1.3 EST与转录表达图谱基因组中仅包括2%左右的序列为编码蛋白质,表达序列标签(Expressed sequence tags,ESTs)和RNA-seq测序可以最有效率的进行基因识别。构建生物特定组织、器官或细胞的cDNA文库并进行大规模EST测序和RNA-seq测序分析,能直接获得大量的功能基因结构及表达特征,并以此来构建各种组织器官的基因表达谱和对基因组结构和功能进行注释。1996年,Le Provost等[31]首次采用泌乳期的山羊乳腺组织构建了cDNA文库,经过筛选对其中的435个cDNA克隆进行EST测序,确认了77个与山羊泌乳有关的基因或者蛋白。2000年,Le Provost等[32]进一步采用图位克隆的技术,结合EST测序和细胞遗传定位技术鉴定了25个可能与产奶性状有关的新的基因,其中6个定位在牛的产奶QTL区域。
RNA-seq技术与生物信息学的快速发展,为理解基因组结构和基因功能奠定了基础。Dong等[15]对云南黑山羊不同组织(肝脏、心脏、肺、肾脏、脾脏、淋巴结、前脑皮层、肌肉、膀胱和卵巢)的mRNA进行了转录组测序,为基因功能注释奠定了坚实的基础。不同组织、细胞的非编码RNA的检测研究,如miRNA(乳腺[33]、皮肤毛囊[34-35]、卵巢[36]、垂体[37]、真皮乳头细胞[38],以及背最长肌[39])、LncRNA(骨骼肌[40]、卵巢[41]和毛囊[42])等的分析研究,也为精细山羊基因组的功能结构、调控元件和基因功能注释提供了数据支持。
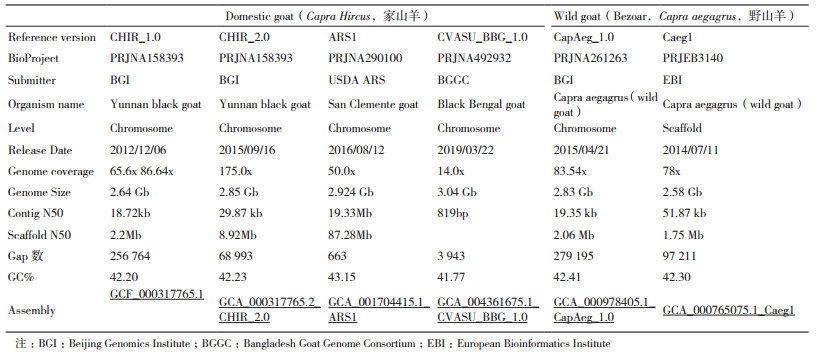
1.4 参考基因组组装2010年3月,国际山羊基因组合作联盟(International goat genome consortium,IGGC)在中国深圳正式成立,由中国科学院昆明动物所、深圳华大基因和内蒙古农业大学等10多个国家的20个科研机构或组织参与,旨在通过国际间的交流合作,加快山羊基因组图谱构建、山羊遗传多样性、环境适应基础和分子育种等方面的研究进展[43]。通过各个研究机构的合作努力和不同的技术方法,先后构建了家山羊参考基因组(CHIR_1.0、CHIR_2.0、ARS1和CVASU_BBG_1.0)和野山羊参考基因组(CapAeg_1.0和Caeg1),为加快山羊的分子生物学研究和今后的基因组选择育种奠定了基础。
1.4.1 家山羊参考基因组(CHIR_1.0与CHIR_2.0)2013年,Dong等[15]利用Illumina测序和光学图谱(Optical mapping)技术以及Fosmid和辐射杂种嵌板技术的数据对云南黑山羊进行基因组从头组装和染色体定位。对云南黑山羊母羊采用双末端测序,构建了7个不同大小片段文库用于基因组测序,共产生191.5 Gb高质量数据。首先,由17-kmer推算和c-value计算山羊的基因组大小,约为2.92 Gb。其次,利用SOAPdenovo软件经过初步组装后的contig N50为18 kb;scaffold N50为2.2 Mb。最后,利用Fosmid和Optical mapping技术方法辅助构建Super-scaffold,获得最终的super-scaffold,获得2.66 Gb大小的参考基因组(CHIR_1.0),组装出的基因组序列占预测基因组大小的92%(2.92 Gb),其Scaffolds N50的大小为18 Mb,无法定位到染色体的super-scaffold归类为chromosome U[15]。此外,利用RH技术对山羊第1号染色体构建了高密度SNP标记的辐射杂种图谱,并与Optical mapping数据组装的长超级支架(Super-scaffold)进行了比对,成功证明了山羊序列的组装质量的可靠性[29]。山羊基因组中含有大量重复序列,约占基因组42.2%。使用从头注释、基于人和牛的基因同源注释和基因预测,总共注释出山羊蛋白编码基因有22 175个,平均转录本长度为29 969 bp,CDS平均长度为1 385 bp,每个基因平均含有8个外显子,每个外显子的平均长度为168 bp,内含子平均长度为3 956 bp。随后,研究人员进一步通过增加Illunima测序数据,对参考基因组CHIR_1.0进行了的一些修正,并利用辐射杂交技术修正了一些scaffold的方向和顺序以及挂载了CHIR_1.0未能成功挂载的scaffold[29]。通过一系列的组装优化工作,最终获得了山羊的基因组序列大小2.85 Gb,contig N50的长度为29.87 kp,scaffold的N50长度为8.92 Mb,其中染色体的GC含量为40.73%,;在使用CHIR_1.0为模板挂载染色体后,同样使用了野山羊染色体和绵羊染色体的作为模板挂载了剩余部分中未成功定位的scaffold,最终在CHIR_2.0中的scaffold序列中能成功挂载到山羊染色体上的序列占总序列的93.2%[44]。总的来说,相较于CHIR_1.0版本的基因组,CHIR_2.0在基因组完整性、功能注释等方面都有较大的提升,极大地促进了山羊遗传变异检测和功能基因定位的研究工作。
1.4.2 家山羊参考基因组(ARS1)2017年,Bickhart等[22]首先利用Illumina的Goat SNP50K芯片从96头山羊(6个品种)中,筛选出基因型纯和度最高的候选个体用来进行基因组从头组装(San clemente)。第一步,用Celera Assembler PacBio corrected Reads流程对Pacbio技术的465个SMRT cell产生的long-read,覆盖深度达69X的194 Gb基因组数据进行初步组装,共获得3 074个contig(2.63 G),其中N50为4.159 Mb。第二步,基于Irys optical mapping技术对其雄性后代测序产生的256 Gb光学图谱数据,并利用IrysView软件构建scaffold,组装产生了842个scaffold,其中,scaffold N50为13.408 Mb(最长的scaffold为66.728 Mb),contig N50为10.858 Mb。第三步,基于PacBio和光学图谱组装的结果,构建Hi-C文库并物理方法打断成300-500 bp大小,双末端(PE101),共产生115 Mb reads的数据量,调用Lachesis软件包,整合PacBio-Irys-PGA(PBIP),获得Scaffold N50为87.347 Mb较为完美的组装结果。第四步,利用Illumina技术,构建PE251测序,获得23X的基因组数据,用来进行一致校正和最后的补洞。最后,利用Kraken v0.10.5去除有病毒和细菌污染的序列,去掉有NCBI vector污染的序列,获得最终的2.924 Gb大小的参考基因组图谱ARS1,包含31个scaffold,663个gap区和680条contig。此外,利用6个组织(大多和脑组织相关)RNA-seq测序数据、13个SRA下载数据,用PASA软件将stringtie、cufflinks和Trinity分析结果整合在一起;用exonerate和tblastn软件比对到几个近缘物种的Ensembl基因集上,获得同源预测基因集;用Braker1做Ab initio预测;CHIR_1.0版本的注释基因集;最后,用EVM+PASA把以上4种数据整合成一个最终的基因集(设置的权重为RNAseq > cDNA/protein > ab initio gene predictions)。此版本基因组是目前组装结果最好的山羊参考基因组,相应的组装策略和技术为其他物种的参考基因组提供了参考,如最新获得水牛基因组组装就采用相似的方法[45]。
1.4.3 家山羊参考基因组(CVASU_BBG_1.0)2019年,Siddiki等采用Illumina测序平台对孟加拉黑山羊进行深度为14 X的150 bp双末端测序,利用ABySS v.2.1.5组装软件初步获得3 294 295个contigs(最小contig大小为200 bp)[46-47];进一步利用ABACAS v.1.3.1组装流程与参考基因组ARS1比较[48],进行从头组装基因组的排列、排序和定向,最终获得了基因组大小为3.04 Gb的孟加拉黑山羊参考基因组(CVASU_BBG_1.0);BUSCO评估基因组的完整性为82.5%[49],基因注释共发现了26 458个基因[50]。孟加拉黑山羊的基因组组装结果为今后深入研究其种群遗传结构、遗传多样性,评估该山羊品种的未来育种潜力奠定了坚实的基础[51]。该研究中利用Illumina短读长数据进行初步组装[47];随后与参考基因组精细图谱(ARS1)比较,利用ABACAS等组装进行基因组序列的排序和定向研究,为今后不同山羊品种的参考基因组组装和进行山羊的泛基因组研究提供了可行性参考。
1.4.4 野山羊参考基因组(CapAeg_1.0)2015年,Dong等[44]采用家山羊CHIR_1.0的DNA文库构建方法对一只雄性野山羊进行测序,基于Illumina Hiseq 2000测序平台共获得了381.50 Gb大小的基因组数据;使用SOAPdenovo软件初步组装获得了野山羊基因组序列;随后,基于野山羊与家山羊基因组的共线性关系,使用LASTZ软件与家山羊参考基因组比对信息,构建了野山羊常染色体基因组。为进一步构建野山羊Y染色体基因组数据,首先利用BLAT软件将常染色体组装中未锚定位置的Scaffolds与家牛Y染色体(家牛Btau_4.6.1的NC_016145.1染色体)参考基因组进行比对;反过来利用LASTZ软件将家牛Y染色体的contigs比对到野山羊Scaffolds上,通过过滤检验分析,最终获得野山羊参考基因组CapAeg_1.0,其中contig N50为18.97 Kb,scaffold N50为2.06 Mb;Y染色体大小为17.3 Mb,包含79个锚定的scaffolds。为注释野山羊基因组的蛋白编码基因,采用了从头预测,同源蛋白比对,转录组测序数据和序列表达标签信息,注释出了23 217个基因;其中注释到了57个Y染色体基因,包括11个已知的雄性特有基因(Male specific region genes,MSY)。获得了大量的遗传变异信息,其中揭示了ASIP基因的拷贝数变异与家山羊的被毛变化相关。
到目前为止,获得的从头组装的参考基因组共有4个品种的个体,其中以ARS1组装注释结果最好(不同参考基因组详细信息见表 1),这些组装到的基因组在一定上促进了山羊泛基因组的研究,为揭示基因组水平大规模的变异奠定的基因组水平的数据基础。
随着测序技术的不断成熟及测序成本的不断降低,利用高通量测序技术检测山羊全基因组水平的遗传变异逐渐成为可能。此外,随着研究对象样本量和品种数的增加,山羊遗传变异的信息也逐渐增加和丰富,极大了加深了我们对的山羊遗传多样性和环境适应性的理解(http://www.genome.gov/sequencingcosts/)。根据遗传变异形成机制、存在形式以及对基因组结构和表型的影响,可分为以下类型,即单核苷酸多态性、1-50 bp的小片段的插入或缺失、50 bp以上的拷贝数变异以及由位置变化引起的易位或倒位等,详细信息如图 1所示[52]。

2010年,Fontanesi等[53]利用牛-山羊间的微阵列比较基因组杂交(Array comparative genome hybridization,aCGH)技术,首次对山羊基因组拷贝数进行了检测研究,共发现了161个CNVs变异。Liu等[54]利用CaprineSNP50芯片和PennCNV软件对ADAPTmap项目产生的基因组数据进行CNV分布分析,从50个山羊品种的1 023个个体中共获得包含6 286个CNVs的978个区域,约262 Mb(8.96%)。基于SNP芯片检测CNV的研究,扩展了SNP芯片的应用范围,加深了对CNV变异在家畜遗传多样性和经济性状差异的理解,但因为SNP芯片的敏感性等原因,其准确性和可靠性需要进一步验证。
基于全基因组个体重测序的方法,Tosser-Klopp[20]、Dong[44]、Benjelloun[55]、Zhang[56]、Florian[55, 57]、Li[58]、Lee[59]、Kim[60]和Cao[61]等对阿尔卑斯山羊、克里奥山羊、Katjang山羊、Savanna山羊、萨能奶山羊、波尔山羊、澳大利亚野化山羊、澳大利亚绒山羊、野山羊、摩洛哥山羊、辽宁绒山羊、内蒙古绒山羊、雷州山羊、韩国黑山羊、努比亚山羊和云岭黑山羊等进行了2.7-30 X不同深度的全基因组测序;采用全基因组混合池测序方法,Lai[62]、Zhang[63-65]、E[65-66]和Wang[67]等通过对崂山奶山羊、大足黑山羊、太行黑山羊、西藏山羊、内蒙古绒山羊、陕北绒山羊、安哥拉山羊、萨能奶山羊、波尔山羊和贵州小山羊等进行了10-30 X的混合池测序;基于简化基因组测序方法,Song等[68]对西藏班戈山羊和日土山羊、柴达木山羊、南疆绒山羊、内蒙古绒山羊二狼山型及辽宁绒山羊)不同个体进行了外显子测序;Wang等[69]利用RNA-seq技术对内蒙古绒山羊阿尔巴斯型进行了遗传变异检测分析。通过与参考基因组比对(CHIR_1.0、CHIR_2.0和ARS1),检测出大量的SNP、Indel和CNV等遗传变异数据,为今后山羊分子遗传标记的开发和利用以及遗传资源保护奠定了坚实的基础。
目前,随着山羊分子生物学的不断发展及对家畜分子育种的重视,许多研究机构对山羊的环境适应性和表型多样性等方面进行了不同程度的研究,详细信息见表 2。因为测序项目实施的时间不同,所用到的山羊参考基因组信息有所不同,导致山羊遗传变异在基因组上位置信息有所差异,为统一山羊基因组变异的相对位置,国际山羊基因组联盟首先对Goat SNP50K芯片的SNP位置信息与ARS1进行了比较和校正。由于NCBI在2017年逐渐停止对dbSNP和dbVar中的所有非人类生物的支持,目前山羊等物种的基因组变异数据存储在Ensemble数据库中(ftp://ftp.ensembl.org/pub/release-97/variation/gvf/capra_hircus/)。截止到2019年5月8日,以参考基因组ARS1版本的作为参考构建的遗传变异信息,主要包括33 996 708个SNP和Indel,而CNV和SV等的变异信息目前尚未公布。
山羊全基因组重测序研究的主要目标就是通过生物信息学方法检测不同品种特有的选择信号特征,揭示不同品种特异性的遗传基础;其次是构建不同品种的全基因组单倍型图谱,为今后利用低密度芯片进行基因型填充、增加基因组信息的可利用率做基础数据支持;再次是利用全基因组水平的遗传变异信息,针对不同的研究群体和目标对SNPs等遗传变异信息进行过滤和筛选,进而开发不同密度的SNP分型芯片。目前,利用不同品种的基因组遗传变异信息,已经成功设计出了Goat SNP50K芯片[20]和66K目标捕获芯片[21]。
基于全基因组重测序数据,在山羊的高海拔环境适应(EPAS1、EDNRA、SIRT1、PASK、PTPRZ1、NPC1L1和RYR1)[68]、脂肪代谢(ACSL1、LRP1、PLIN4、FASN)、绒用性状(FGF5、PRDM6)[56, 58, 67]、被毛颜色(KITLG、MC1R、ASIP、ATRN、GNAQ、HELLS、MUTED、OSTM1、TRPM7、VPS33A、Ada-mts20,MITF、OCA2、SLC7A11和AHCY)[44, 55, 57]、神经系统发育(ADRA2A、FXR2、HTR3A、CACNA1、CCHD5、ULK1、TMEM132A、SYNDIG1、ERC2和GABRB2)[44, 56]、繁殖性状(NR6A1、STK3、IGF2-BP2、NPTX1、ANKRD17、DPYD、CLRB、PPP3CA,PLCB1,STK3 and HMGA2,PRP1、PRP6、CCNB2、AR、ADCY1、DNMT3B、SMAD2、AMHR2、ERBB2、FGFR1,MAP3K12、SETDB2、CDH26和THEM4)[62, 64-65]、体尺性状(NR6A1、TNFSF13、STIM1、IGF1R)[44, 56]、肉用性状(GDF5、LRP4、HMGXB3、SLC26A2、goat_GLEAN 10018710、SLC-35A3、HIAT1、SASS6和GOAT_ENSBTAP00000044-216)[56]、疾病抗性(HTT、CCR3)[55, 59]、生长性状(CCKAR、IGF1R、MYADM)[44]、免疫系统(ABCC4、PRAME、CD163L1、KIR3DL1、CFH和TRIM5)[44]、精子发生(PRAME)[44]和乳用性状(BTN1A1、RSRC1、SHOX2、VPS13A、VPS13B、VPS13C和RPL3)[44, 56]等遗传基础的解析方面取得了众多研究成果。
4 展望目前,山羊重要经济性状遗传基础的研究正在由候选基因、单一性状的方法向全基因组水平、多性状和多组学等联合分析的方法进行转变。高通量测序技术的进一步发展和新的分析方法的不断涌现,加快了研究人员研究、挖掘全基因组范围内山羊的遗传多样性信息及经济性状相关的分子基础,如Guan等[71]基于共享基因组数据分析山羊酪蛋白基因家族变异的起源与演化过程。山羊大多数经济性状属于数量性状,遗传因素如单碱基突变(SNP)、插入缺失(Indel)、结构变异(SV)和表观遗传修饰调控(甲基化修饰、组蛋白修饰和非编码RNA调控)以及环境和营养因素等均会影响到山羊的表型性状和生产性能。为揭示复杂性状的遗传基础和调控机制,高通量技术下的研究方法主要包括对不同组织器官的差异基因表达的RNA-seq分析、基于不同品种杂交个体的等位基因特异性表达分析、基于全基因组重测序技术的选择性清除分析、复杂性状基因定位的全基因组关联分析、表观遗传调控组蛋白修饰和甲基化分析以及非编码RNA调控的研究以及逐渐在上述技术方法基础上衍生的多组学方法,如RNA-seq + GWAS、WGS + GWAS、eGWAS和BSA + RNA-seq等联合分析进行精确定位的研究方法[71-73]。通过合理的选择研究对象、构建理想的试验群体并适当的组学技术,借助公共数据库基因组信息和生物信息学方法挖掘其潜在的与生产性状相关的基因或基因组区域、影响效应和调控互作机制将是今后的研究重点,也对推动山羊分子育种和基因组选择研究工作具有重要的理论和实践意义。
| [1] |
Zeder MA, Hesse B. The initial domestication of goats(Capra hircus)in the Zagros mountains 10, 000 years ago[J]. Science, 2000, 287(5461): 2254-2257. DOI:10.1126/science.287.5461.2254 |
| [2] |
Daly KG, Maisano Delser P, Mullin VE, et al. Ancient goat genomes reveal mosaic domestication in the Fertile Crescent[J]. Science, 2018, 361(6397): 85-88. DOI:10.1126/science.aas9411 |
| [3] |
Devendra C. Dynamics of goat meat production in extensive systems in Asia:improvement of productivity and transformation of livelihoods[J]. Agrotechnology, 2015, 106(2): 275-277. |
| [4] |
Bertolini F, Servin B, Talenti A, et al. Signatures of selection and environmental adaptation across the goat genome post-domestication[J]. Genetics, Selection, Evolution, 2018, 50(1): 421-444. |
| [5] |
Boettcher PJ, Hoffmann I, Baumung R, et al. Genetic resources and genomics for adaptation of livestock to climate change[J]. Frontiers in Genetics, 2015, 5(461): 107-109. |
| [6] |
Bett RC, S Kosgey I, Bebe B, et al. Breeding goals for the Kenya dual purpose goat. I. model development and application to smallholder production systems[J]. Tropical Animal Health and Production, 2007, 39: 477-492. DOI:10.1007/s11250-007-9015-3 |
| [7] |
Zook JM, Chapman B, Wang J, et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls[J]. Nature Biotechnology, 2014, 32(3): 246-251. DOI:10.1038/nbt.2835 |
| [8] |
Liao PY, H. Lee K. From SNPs to functional polymorphism:The insight into biotechnology applications[J]. Biochemical Engineering Journal, 2010, 49: 149-158. DOI:10.1016/j.bej.2009.12.021 |
| [9] |
Kwok PY, Chen X. Detection of single nucleotide polymorphisms[J]. Current Issues in Molecular Biology, 2003, 5(2): 43-60. |
| [10] |
Davey JW, Hohenlohe PA, Etter PD, et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing[J]. Nature Reviews. Genetics, 2011, 12(7): 499-510. DOI:10.1038/nrg3012 |
| [11] |
Helyar SJ, Hemmer-Hansen J, Bekkevold D, et al. Application of SNPs for population genetics of nonmodel organisms:new opportunities and challenges[J]. Molecular Ecology Resources, 2011, 1: 123-136. |
| [12] |
Elsik CG, Tellam RL, Worley KC, et al. The genome sequence of taurine cattle:a window to ruminant biology and evolution[J]. Science, 2009, 324(5926): 522-528. DOI:10.1126/science.1169588 |
| [13] |
Wade CM, Giulotto E, Sigurdsson S, et al. Genome sequence, comparative analysis, and population genetics of the domestic horse[J]. Science, 2009, 326(5954): 865-867. DOI:10.1126/science.1178158 |
| [14] |
Groenen MA, Archibald AL, Uenishi H, et al. Analyses of pig genomes provide insight into porcine demography and evolution[J]. Nature, 2012, 491(7424): 393-398. DOI:10.1038/nature11622 |
| [15] |
Dong Y, Xie M, Jiang Y, et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat(Capra hircus)[J]. Nature Biotechnology, 2013, 31(2): 135-141. DOI:10.1038/nbt.2478 |
| [16] |
Jiang Y, Xie M, Chen W, et al. The sheep genome illuminates biology of the rumen and lipid metabolism[J]. Science, 2014, 344(6188): 1168-1173. DOI:10.1126/science.1252806 |
| [17] |
Matukumalli LK, Lawley CT, Schnabel RD, et al. Development and characterization of a high density SNP genotyping assay for cattle[J]. PLoS One, 2009, 4(4): 24-36. |
| [18] |
Levy SE, Boone BE. Next-generation sequencing strategies[J]. Cold Spring Harbor Perspectives in Medicine, 2019, 9(7): a025791. DOI:10.1101/cshperspect.a025791 |
| [19] |
Zhang J, Chiodini R, Badr A, et al. The Impact of next-generation sequencing on genomics[J]. Journal of Genetics and Genomics, 2011, 38: 95-109. DOI:10.1016/j.jgg.2011.02.003 |
| [20] |
Tosser-Klopp G, Bardou P, Bouchez O, et al. Design and characterization of a 52K SNP chip for goats[J]. PLoS One, 2014, 9(1): e86227. DOI:10.1371/journal.pone.0086227 |
| [21] |
Qiao X, Su R, Wang Y, et al. Genome-wide target enrichment-aided chip design:a 66 K SNP chip for cashmere goat[J]. Scientific Reports, 2017, 7(1): 8621-8633. DOI:10.1038/s41598-017-09285-z |
| [22] |
Bickhart DM, Rosen BD, Koren S, et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome[J]. Nature Genetics, 2017, 49(4): 643-650. DOI:10.1038/ng.3802 |
| [23] |
Vaiman D, Schibler L, Bourgeois F, et al. A linkage map of the male goats genome[J]. Genetics, 1996, 144: 279-305. |
| [24] |
Schibler L, Vaiman D, Oustry A, et al. Construction and extensive characterization of a goat bacterial artificial chromosome library with threefold genome coverage[J]. Mammalian Genome, 1998, 9(2): 119-124. DOI:10.1007/s003359900701 |
| [25] |
Schibler L, Vaiman D, Oustry A, et al. Comparative gene mapping:a fine-scale survey of chromosome rearrangements between ruminants and humans[J]. Genome Research, 1998, 8(9): 901-915. DOI:10.1101/gr.8.9.901 |
| [26] |
Maddox JF. A presentation of the differences between the sheep and goat genetic maps[J]. Genetics, Selection, Evolution, 2005, 37(1): S1-S10. |
| [27] |
Goss SJ, Harris H. New method for mapping genes in human chromosomes[J]. Nature, 1975, 255(5511): 680-684. DOI:10.1038/255680a0 |
| [28] |
Du XY, Womack EJ, Owens EK, et al. A whole-genome radiation hybrid panel for goat[J]. Small Ruminant Research, 2012, 105: 114-116. DOI:10.1016/j.smallrumres.2011.11.023 |
| [29] |
Du XY, Servin B, Womack JE, et al. An update of the goat genome assembly using dense radiation hybrid maps allows detailed analysis of evolutionary rearrangements in Bovidae[J]. BMC Genomics, 2014, 15(625): 625-640. |
| [30] |
Schibler L, Di Meo GP, Cribiu EP, et al. Molecular cytogenetics and comparative mapping in goats(Capra hircus, 2n = 60)[J]. Cytogenetic and Genome Research, 2009, 126(1-2): 77-85. DOI:10.1159/000245908 |
| [31] |
Le Provost F, Lepingle A, Martin P. A survey of the goat genome transcribed in the lactating mammary gland[J]. Mammalian Genome, 1996, 7(9): 657-666. DOI:10.1007/s003359900201 |
| [32] |
Le Provost F, Schibler L, Oustry-Vaiman A, et al. Cytogenetic mapping of 25 goat mammary gland expressed sequence tags(ESTs)[J]. Genetics, Selection, Evolution, 2000, 32(3): 311-320. DOI:10.1186/1297-9686-32-3-311 |
| [33] |
Mobuchon L, Marthey S, Boussaha M, et al. Annotation of the goat genome using next generation sequencing of microRNA expressed by the lactating mammary gland:comparison of three approaches[J]. BMC Genomics, 2015, 16(285): 1471-1486. |
| [34] |
Liu Y, Wang LL, Li XY, et al. High-throughput sequencing of hair follicle development-related micrornas in cashmere goat at various fetal periods[J]. Saudi Journal of Biological Sciences, 2018, 25(7): 1494-1508. DOI:10.1016/j.sjbs.2017.12.009 |
| [35] |
Liu ZH, Xiao HM, Li HP, et al. Identification of conserved and novel microRNAs in cashmere goat skin by deep sequencing[J]. PLoS One, 2012, 7(12): e50001. DOI:10.1371/journal.pone.0050001 |
| [36] |
Ling YH, Ren CH, Guo XF, et al. Identification and characterization of microRNAs in the ovaries of multiple and uniparous goats(Capra hircus)during follicular phase[J]. BMC Genomics, 2014, 15(339): 1471-2164. |
| [37] |
Ye J, Yao ZQ, Si WY, et al. Identification and characterization of microRNAs in the pituitary of pubescent goats[J]. Reproductive Biology and Endocrinology, 2018, 16(1): 370-379. |
| [38] |
Ma S, Wang Y, Zhou GX, et al. Synchronous profiling and analysis of mRNAs and ncRNAs in the dermal papilla cells from cashmere goats[J]. BMC Genomics, 2019, 20(1): 5861-5875. |
| [39] |
Guo JZ, Zhao W, Zhan SY, et al. Identification and expression profiling of miRNAome in goat longissimus dorsi muscle from prenatal stages to a neonatal stage[J]. PLoS One, 2016, 11(10): e0165764. DOI:10.1371/journal.pone.0165764 |
| [40] |
Zhan SY, Dong Y, Zhao W, et al. Genome-wide identification and characterization of long non-coding RNAs in developmental skeletal muscle of fetal goat[J]. BMC Genomics, 2016, 17(666): 3009-3018. |
| [41] |
Liu Y, Qi B, Xie J, et al. Filtered reproductive long non-coding RNAs by genome-wide analyses of goat ovary at different estrus periods[J]. BMC Genomics, 2018, 19(1): 5268-5280. |
| [42] |
Zhou GX, Kang DJ, Ma S, et al. Integrative analysis reveals ncRNA-mediated molecular regulatory network driving secondary hair follicle regression in cashmere goats[J]. BMC Genomics, 2018, 19(1): 222-237. DOI:10.1186/s12864-018-4603-3 |
| [43] |
Tosser-Klopp G, Bardou P, Cabau C, et al. Goat genome assembly, availability of an international 50K SNP chip and RH panel: an update of the international goat genome consortium projects[C]. San Diego, CA: Plant and Animal Genome, 2012.
|
| [44] |
Dong Y, Zhang XL, Xie M, et al. Reference genome of wild goat(Capra aegagrus)and sequencing of goat breeds provide insight into genic basis of goat domestication[J]. BMC Genomics, 2015, 16(431): 431-441. |
| [45] |
Low WY, Tearle R, Bickhart DM, et al. Chromosome-level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity[J]. Nature Communications, 2019, 10(1): 260-270. DOI:10.1038/s41467-018-08260-0 |
| [46] |
Simpson JT, Wong K, Jackman SD, et al. ABySS:a parallel assembler for short read sequence data[J]. Genome Research, 2009, 19(6): 1117-1123. DOI:10.1101/gr.089532.108 |
| [47] |
Paszkiewicz K, Studholme DJ. De novo assembly of short sequence reads[J]. Briefings in Bioinformatics, 2010, 11(5): 457-472. DOI:10.1093/bib/bbq020 |
| [48] |
Assefa S, Keane TM, Otto TD, et al. ABACAS:algorithm-based automatic contiguation of assembled sequences[J]. Bioinformatics, 2009, 25(15): 1968-1969. DOI:10.1093/bioinformatics/btp347 |
| [49] |
Seppey M, Manni M, Zdobnov EM. BUSCO:assessing genome assembly and annotation completeness[J]. Methods in Molecular Biology, 2019, 9170-9173. |
| [50] |
Siddiki AZ, Baten A, Billah M, et al. The genome of the Black Bengal goat(Capra hircus)[J]. BMC Research Notes, 2019, 12(1): 362-364. DOI:10.1186/s13104-019-4400-3 |
| [51] |
Faruque S, Chowdhury SA, Siddiquee NU, et al. Performance and genetic parameters of economically important traits of Black Bengal goat[J]. Journal of the Bangladesh Agricultural University, 2010, 8: 67-78. |
| [52] |
Pollex RL, Hegele RA. Copy number variation in the human genome and its implications for cardiovascular disease[J]. Circulation, 2007, 115(24): 3130-3138. DOI:10.1161/CIRCULATIONAHA.106.677591 |
| [53] |
Fontanesi L, Martelli PL, Beretti F, et al. An initial comparative map of copy number variations in the goat(Capra hircus)genome[J]. BMC Genomics, 2010, 11(639): 1471-2164. |
| [54] |
Liu M, Zhou Y, Rosen BD, et al. Diversity of copy number variation in the worldwide goat population[J]. Heredity, 2018, 6(10): 150-161. |
| [55] |
Benjelloun B, Alberto FJ, Streeter I, et al. Characterizing neutral genomic diversity and selection signatures in indigenous populations of Moroccan goats(Capra hircus)using WGS data[J]. Frontiers in Genetics, 2015, 6(107): 107-121. |
| [56] |
Zhang B, Chang L, Lan YX, et al. Genome-wide definition of selective sweeps reveals molecular evidence of trait-driven domestication among elite goat(Capra species)breeds for the production of dairy, cashmere, and meat[J]. Gigascience, 2018, 7(12). DOI:10.1093/gigascience/giy105 |
| [57] |
Alberto FJ, Boyer F, Orozco-Terwengel P, et al. Convergent genomic signatures of domestication in sheep and goats[J]. Nature Communications, 2018, 9(1): 813-822. DOI:10.1038/s41467-018-03206-y |
| [58] |
Li XK, Su R, Wan WT, et al. Identification of selection signals by large-scale whole-genome resequencing of cashmere goats[J]. Scientific Reports, 2017, 7(1): 142-151. DOI:10.1038/s41598-017-00209-5 |
| [59] |
Lee W, Ahn S, Taye M, et al. Detecting positive selection of Korean native goat populations using next-generation sequencing[J]. Molecules and Cells, 2016, 39(12): 862-868. DOI:10.14348/molcells.2016.0219 |
| [60] |
Kim JY, Jeong S, Kim KH, et al. Discovery of genomic characteristics and selection signatures in Korean indigenous goats through comparison of 10 goat breeds[J]. Frontiers in Genetics, 2019, 10(699): 699-716. |
| [61] |
Cao YH, Xu H, Li R, et al. Genetic basis of phenotypic differences between Chinese Yunling black goats and Nubian goats revealed by allele-specific expression in their F1 hybrids[J]. Frontiers in Genetics, 2019, 10(145): 145-156. |
| [62] |
Lai FN, Zhai HL, Cheng M, et al. Whole-genome scanning for the litter size trait associated genes and SNPs under selection in dairy goat(Capra hircus)[J]. Scientific Reports, 2016, 6. DOI:10.1038/srep38096 |
| [63] |
Zhang SL, Cao XY, Li Y, et al. Detection of polled intersex syndrome(PIS)and its effect on phenotypic traits in goats[J]. Animal Biotechnology, 2019, 14: 1-5. |
| [64] |
Zhang RQ, Wang JJ, Zhang T, et al. Copy-number variation in goat genome sequence:A comparative analysis of the different litter size trait groups[J]. Gene, 2019, 696: 40-46. DOI:10.1016/j.gene.2019.02.027 |
| [65] |
E GX, Zhao YJ, Huang YF. Selection signatures of litter size in Dazu black goats based on a whole genome sequencing mixed pools strategy[J]. Molecular Biology Reports, 2019, 7(10): 4904-4911. |
| [66] |
E GX, Jin ML, Zhao YJ, et al. Genome-wide analysis of Chongqing native intersexual goats using next-generation sequencing[J]. 3 Biotech, 2019, 9(3): 99-112. |
| [67] |
Wang XL, Liu J, Zhou GX, et al. Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits[J]. Scientific Reports, 2016, 6(38932): 932-941. |
| [68] |
Song S, Yao N, Yang M, et al. Exome sequencing reveals genetic differentiation due to high-altitude adaptation in the Tibetan cash-mere goat(Capra hircus)[J]. BMC Genomics, 2016, 17(122): 2449-2461. |
| [69] |
Wang LL, Zhang YJ, Zhao M, et al. SNP discovery from transcrip-tome of cashmere goat skin[J]. Asian-Australasian Journal of Animal Sciences, 2015, 28(9): 1235-1243. DOI:10.5713/ajas.15.0172 |
| [70] |
Guan DL, Marmol-Sanchez E, Cardoso TF, et al. Genomic analysis of the origins of extant casein variation in goats[J]. Journal of Dairy Science, 2019, 102(6): 5230-5241. DOI:10.3168/jds.2018-15281 |
| [71] |
Hawe JS, Theis FJ, Heinig M. Inferring interaction networks from multi-omics data[J]. Frontiers in Genetics, 2019, 10(535): 535-548. |
| [72] |
Teng C, Du DZ, Xiao L, et al. Mapping and identifying a candidate gene(Bnmfs)for female-male sterility through whole-genome resequencing and RNA-Seq in rapeseed(Brassica napus L.)[J]. Frontiers in Plant Science, 2017, 8(2086): 2086-2088. |
| [73] |
Sun YV, Hu YJ. Integrative analysis of multi-omics data for discovery and functional studies of complex human diseases[J]. Advances in Genetics, 2016, 93: 147-190. DOI:10.1016/bs.adgen.2015.11.004 |