




构建重组质粒是现代分子生物学中十分重要的技术。传统的质粒构建多采用限制性核酸内切酶和DNA连接酶的方式[1],但这种方法常依赖酶切位点,酶切耗时较长还可能会引入一段不需要的序列,另外限制性核酸内切酶的价格也较为昂贵。近几年以来,无缝克隆的方法发展迅猛[2-5],相比较传统方法,无缝克隆的方法不受酶切位点限制而且操作简便快速。
目前市场上无缝克隆试剂盒种类繁多,如NEB公司的NEBuilder试剂盒、Thermo公司的GeneArt试剂盒、CloneTech公司的In-Fusion试剂盒等。其采用的无缝克隆技术和原理各有不同,共同的特点是成本相对较高,每次反应大约需要18-36美元[6]。Zhang等[7]所建立的SLiCE(Seamless ligation cloning extract)方法是一种利用大肠杆菌细胞内同源重组相关酶进行DNA克隆的新方法,其机理在于不依赖于RecA-的片段重组[8-12]。Motohashi[13]从成本角度进一步优化了SLiCE技术,选用RecA-实验菌株E.col JM109提取细胞裂解液,并优化了裂解液配方降低了裂解成本,使基于SLiCE技术的反应折合人民币仅仅0.4元一次[14]。相比较于市面上的无缝克隆技术,SLiCE技术为目前最便宜的无缝克隆技术[14]。
虽然SLiCE反应能实现较为经济的无缝克隆,但是SLiCE反应产物的转化依赖于高转化效率的感受态细胞,例如Zhang等[7]采用1×1010 CFU/μg电转化效率的感受态细胞和1×109 CFU/μg的化学转化效率的感受态细胞,Motohashi[13]则采用的是1×108 CFU/μg化学转化效率的感受态细胞。商业化的高感受态细胞价格不菲,但自制高感受态细胞工序较为繁琐,且转化效率往往不稳定,因此很多无缝克隆方法建立时都考虑到了感受态细胞效率这个因素[6, 14]。
对于无缝克隆的高感受态细胞依赖问题,Zhang等[7]将λ噬菌体Red/ET重组系统导入到DH108菌株用于SLiCE反应,通过提高克隆子形成率从而提高克隆效率[14],由于菌株不易获得使得该方法推广到各个实验室有一定困难;王广珺等[15]在使用T4聚合酶进行无缝克隆的基础上,通过添加退火缓冲液进行退火处理,将重组效率提高了10倍之多。
据最新报道体内细胞进行核酸片段重组是基于核酸外切酶[16],对于采用细胞裂解液进行无缝克隆的SLiCE而言,我们推测采用退火处理[15]的方式也有可能提高SLiCE的重组效率。此外,电转化法很容易获得很高的转化效率,但此前无缝克隆采用电转化鲜有成功的例子,我们推测很可能与无缝克隆反应带来杂质有关,如SLiCE反应就有反应缓冲液,因此本文对SLiCE反应产物进行了纯化后再实施电转化,以期待获得更高的克隆效率。
1 材料与方法 1.1 材料 1.1.1 菌株和质粒本研究采用的菌株大肠杆菌(Escherichia coli)JM109、DH5α、MG1655以及质粒pUC19等均为实验室保存。
1.1.2 培养基LB培养基:1%胰蛋白胨,0.5%酵母提取物,0.5% NaCl;2×YT培养基:1.6%胰蛋白胨,1%酵母提取物,0.5% NaCl;SOC培养基:2%胰蛋白胨,0.5%酵母提取物,10 mmol/L NaCl,2.5 mmol/L KCl,10 mmol/L MgCl2,10 mmol/L MgSO4,20 mmol/L葡萄糖。
1.1.3 酶和试剂高保真酶:PrimeSTAR Max DNA PoLymerase,大连TaKaRa产品;Taq聚合酶:2×Taq Master Mix(Dye Plus),南京Vazyme产品;限制性核酸内切酶:EcoR Ⅰ和BamH Ⅰ,大连TaKaRa产品;裂解缓冲液:3%(W/V)Triton X-100,50 mmol/L Tris-HCl(pH 8.0)[17];SLiCE buffer(10×):500 mmol/L Tris-HCl,pH 7.5,100 mmol/L MgCl2,10 mmol/L ATP,10 mmol/L二硫苏糖醇,用0.22 μm滤膜过滤,以40 μL体积分装到PCR管中,放置-20℃保存;10×退火缓冲液:0.1 mol/L Tris-HCl,pH 8.0,1 mol/L NaCl,10 mmol/L EDTA;硅藻土悬浊液:称取1 g硅藻土(Celite545,Fluka),用5 mL蒸馏水悬浮并轻轻倒入已装有45 mL蒸馏水的Falcone管中,4 min后回收悬浊液,再经1 300×g离心1 min,弃上清液。离心所得的颗粒用1 mL蒸馏水悬浮,经高压灭菌后4℃保存备用[18]。
1.1.4 仪器与设备MyCycler PCR仪,美国Bio-Rad公司产品;DYY-6C型电泳仪,北京是六一仪器厂产品;凝胶成像仪、金属浴恒温仪,美国Major Science公司产品;Centrifuge 5417R、Centrifuge 5804R离心机,美国Eppendorf公司产品。
1.2 方法 1.2.1 插入DNA片段和线性化载体DNA的制备有两种方式都可将质粒变成线性化载体,其具体做法如下:
1.2.1.1 双酶切法制备线性化载体以来源于E.col MG1655的可溶性吡啶核苷酸转氢酶(Soluble Pyridine Nucleotide Transhydrogenase,udhA)的基因片段udhA(1 401 bp)作为插入基因,pUC19质粒作为线性化载体的模板。插入DNA片段采用表 1的udhA-F和udhA-R引物进行扩增,采用限制性内切酶EcoR Ⅰ和BamH Ⅰ对pUC19质粒进行切割以获得双酶切线性化载体。随后DNA片段都经过乙醇沉淀和胶回收试剂盒(OMEGA)纯化。

采用Linearized pUC-F和Linearized pUC-R引物,以pUC19为模板,采用高保真酶进行PCR,PCR条件为:98℃预变性30 s;98℃变性10 s,50℃退火15 s,72℃延伸30 s,共30个循环;最后72℃延伸5 min。对应的插入DNA片段则采用表 1的Linearized udhA-F和Linearized udhA-R引物进行扩增。插入DNA和载体DNA的同源区域大小设计为19 bp。
1.2.2 SLiCE提取物的制备及反应参考Okegawa等[14]的方法制备SLiCE提取物:从LB平板上挑取JM109单菌落于LB培养基(1 mL)中,放置摇床37℃、200 r/min过夜培养。然后转到装有50 mL 2×YT培养基的摇瓶中,37℃,200 r/min培养至OD600到3.0(对数生长后期,约3.5 h),随后在4℃及5 000×g条件下离心10 min,弃去上清液,加入50 mL预冷的无菌水洗涤,再一次在4℃及5 000×g条件下离心5 min,弃去上清液。加入1.2 mL裂解缓冲液轻轻的重悬,室温放置10 min。细胞裂解产物在16 000×g,4℃离心2 min,上清液转移到1.5 mL管与等体积的冰浴的80%(V/V)甘油混合。以上制备所得的SLiCE提取物以40 μL每管装到0.2 mL PCR管,干冰速冻后放置于-80℃冰箱长期保存,如3个月内使用也可放置在-20℃冰箱。
SLiCE反应溶液由以下成分组成:线性化载体DNA,适量的插入DNA片段,1 μL 10×SLiCE buffer,1 μL SLiCE提取物,加入无菌水使反应体系达到10 μL。将SLiCE反应混合物放于金属浴恒温仪在37℃反应30 min,随后进行转化。
1.2.3 SLiCE反应产物的转化选用了两种方式制备感受态细胞,并用1 μL浓度为1 ng/μL的pUC19质粒测定了感受态细胞的转化效率。采用Inoue法[19]制备感受态细胞,测得转化效率约为1×107 CFU/μg;采用传统的CaCl2法制备感受态细胞,测得转化效率为1×105-1×106 CFU/μg。
SLiCE反应产物化学转化方法:将100 μL感受态细胞放置冰上解冻,加入10 μL的SLiCE反应产物,轻轻混匀,置于冰水浴放置30 min,然后放置在42℃水浴锅中90 s后,迅速放回冰上停留2 min,随后加入预热至37℃的890 μL SOC培养基,混匀,转移到10 mL离心管,于37℃、200 r/min的摇床中培养1 h。将菌液于离心机12 000 r/min室温离心1 min,弃去上清800 μL,用剩余的上清液重悬沉淀,全部涂布于含100 μg/mL氨苄青霉素的LB平板上,37℃培养12-16 h。
1.2.4 克隆子验证挑取转化子作为模板,采用Taq聚合酶和验证引物V-pUC-udhA-F和V-pUC-udhA-R(表 1)进行菌落PCR。PCR条件为95℃预变性5 min;95℃变性10 s,53℃退火10 s,72℃延伸1 min,共30个循环;最后72℃延伸10 min。采用0.8%琼脂糖凝胶在100 V电压下对PCR产物进行电泳分析,如果条带大小为1 982 bp表明该转化子为克隆子,若条带大小为552 bp,则为未被酶切的质粒形成的转化子,判定为假阳性。根据电泳结果统计克隆子数,根据公式计算重组效率:

在Okegawa等[14]的研究结果发现,当插入DNA片段与载体的摩尔比在10:1时,SLiCE反应后克隆效率最高。基于此我们在对DNA浓度进行优化前先将插入DNA片段与载体的摩尔比确定为10:1。
为明确DNA浓度对SLiCE反应效率的影响,在确定插入DNA与线性化载体摩尔比为10:1的条件下,设定线性化载体浓度为10 ng、50 ng和100 ng,考察线性化载体浓度对SLiCE反应的影响。将插入DNA片段(基因片段)和线性化载体在37℃下进行SLiCE反应30 min,随后将SLiCE反应产物全部转化至感受态细胞(转化pUC19质粒效率为1.16×107 CFU/μg),进行克隆子验证并计算重组效率。
1.2.5.2 退火方式对SLiCE反应的影响采用两种不同退火方式考察退火方式对SLiCE反应的影响:
(1)先退火处理,再进行SLiCE反应:在DNA片段混合液(含50 ng线性化载体,插入DNA与线性化载体摩尔比为10:1)中加入1 μL 10×退火缓冲液,用超纯水补充至总体积为8 μL,进行以下退火程序:75℃ 10 min,每1 min降低1℃,至37℃。随后加入1 μL 10×SLiCE buffer与1 μL SLiCE提取液在37℃下反应30 min;
(2)先进行SLiCE反应,后退火处理:DNA片段混合液先加入1 μL 10×SLiCE buffer与1 μL SLiCE提取物并用ddH2O补充至总体积9 μL,37℃下进行SLiCE反应30 min,随后加入1 μL 10×退火缓冲液进行退火程序。将SLiCE反应产物全部转化至感受态细胞(转化pUC19质粒效率为1.16×107 CFU/μg),进行克隆子验证并计算重组效率。
1.2.5.3 电转化法对SLiCE反应的影响相对于化学转化法,电转化法转化效率更高,但是用于无缝克隆效果并不理想。我们认为其原因可能是未将无缝克隆反应后的溶液进行纯化。
(1)SLiCE反应产物的纯化将DNA片段混合液(含50 ng线性化载体,插入DNA与线性化载体摩尔比为10:1)在37℃下进行SLiCE反应30 min,随后加入退火缓冲液进行退火程序,再利用硅藻土悬浊液将SLiCE反应产物进行纯化[20]:将SLiCE反应后的溶液先用3倍体积的6 mol/L NaI混合室温放置2-5 min,加入到约20 μL硅藻土悬浊液中,混匀,冰上放置15 min,期间每分钟混匀1次。12 000 r/min离心5 min,弃上清液,沉淀用70%乙醇悬浮。再次12 000 r/min离心5 min,弃上清。沉淀在室温条件下自然干燥数分钟后,加入经70℃预热的30 μL的超纯水,室温静置2-5 min,期间每分钟混匀1次。最后12 000 r/min离心5 min,在尽量不要吸到硅藻土条件下取上清液,于-20℃保存备用。
(2)电转化感受态细胞制备及电转化从LB平板上挑取E.col DH5α单菌落于2 mL的LB培养基中,放置摇床37℃、200 r/min过夜培养后,取1 mL菌液接种到50 mL LB培养基中,一共接种两瓶,放置摇床37℃、200 r/min培养至OD600到0.3-0.4。菌液放置冰上预冷30 min,随后在4℃、6 000 r/min离心5 min,弃上清,用40 mL预冷的灭菌后的超纯水重悬,再次离心,收集两管的菌体合并后重悬至一管,然后离心弃上清,用预冷的灭菌后的超纯水重悬至总体积约为240 μL。取80 μL菌液与10 μL纯化后的SLiCE反应液于冰上预混,转移至电极杯,放至电转化仪(Bio-Rad)调电压为2.5 kv进行电击,电击时间约4.9 ms,立即加入200 μL预热至37℃的SOC培养基,使用移液器轻轻吹打混合。转移所有菌液到10 mL离心管,补加SOC培养基至1 mL,37℃,200 r/min复苏1 h。所有菌液涂布于含100 μg/mL氨苄青霉素的LB平板上,37℃培养12-16 h后进行克隆子验证并计算重组效率。
1.2.5.4 感受态细胞的转化效率对SLiCE的影响分别采用1×105、1×106和1×107 CFU/μg转化效率的感受态细胞转化SLiCE反应,以确定感受态细胞转化效率对SLiCE的影响。SLiCE反应采用10 ng的线性化载体,插入DNA与线性化载体摩尔比为10:1,于37℃反应30 min,随后化转到相应转化效率的感受态细胞中,随后进行克隆子验证并计算重组效率。
2 结果 2.1 DNA浓度对SLiCE反应的影响我们将插入DNA片段的浓度确定为载体摩尔浓度的10倍,对SLiCE反应所需的线性化载体的量进行了优化,结果如表 2所示。

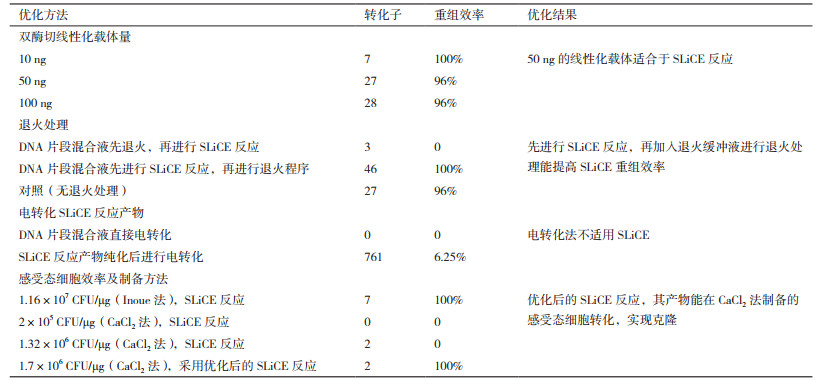
当线性化载体的量从10 ng提升到50 ng时,转化子数目有了很大幅度的提升,提高了近4倍,当线性化载体的量从50 ng提升到100 ng后,转化子数目变化不大。挑取转化子进行PCR验证实验发现,提高线性化载体的量后重组效率仍保持在很高的水平(图 1)。

|
| M:DL 5000 marker;C:以pUC19质粒为模板进行PCR作为阴性对照;1-16:挑取的16个转化子进行PCR验证 图 1 菌落PCR验证SLiCE克隆结果 |
在线性化载体为50 ng,插入DNA与线性化载体摩尔比为10:1的条件下,SLiCE优化实验中设置了不同的退火方式,结果如表 3所示。

相对于对照,先进行SLiCE反应,后退火处理,转化子数目提高了2倍,重组效率也达到了100%。而先退火处理,再进行SLiCE反应,仅产生3个转化子,且经过菌落PCR验证没有发现克隆子,表明先进行SLiCE反应,后退火处理的方式能够提高SLiCE的克隆效率。
线性化载体片段制备有两种方式,除了上述酶切方式制备线性化载体片段外,通过PCR方式制备线性化载体片段具有不依赖限制性内切酶位点的特点也广泛采用。我们对50 ng的PCR线性化载体,插入DNA与线性化载体摩尔比为10:1,采用先进行SLiCE反应,后退火处理的方式进行了反应,结果发现,转化子数目为52,挑取克隆子进行PCR验证,重组成功率为100%。相比酶切方式制备线性化载体片段,获得克隆子数目更多。因此,PCR法制备线性化载体不仅适用于优化后的SLiCE方案,且能够提升SLiCE的克隆效率。
2.3 电转化对于SLiCE反应的影响先对SLiCE反应产物进行纯化,再采用电转化法转化至效率为3.26×109 CFU/μg的电转化感受态细胞,获得761个的转化子,但对挑取的20个单菌落进行PCR验证后仅有一个为克隆子(图 2)。结果表明电转化法虽然可以提高转化DNA片段转化到胞内的能力,但不管是酶切方式还是PCR方式获得线性化载体的方法都会残留一些完整的质粒,这些也带有氨苄抗性的质粒也会高效转化到菌体中形成转化子,产生假阳性,这样就加大了后续的筛选的工作量。在转化前加入DpnⅠ酶有可能减少假阳性,但会增加额外成本和步骤,对于SLiCE无缝克隆的经济性有一定影响,因此也不建议电转化方式用于SLiCE无缝克隆。

|
| DL 5000 marker;C:以pUC19质粒为模板进行PCR作为阴性对照;1-20:挑取的20个克隆子进行PCR验证 图 2 菌落PCR验证电转化方式下的SLiCE克隆结果 |
为克服对感受态细胞的依赖,首先测试了不同转化效率的感受态细胞对SLiCE的影响,结果如表 4所示。
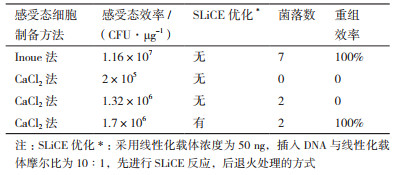
当采用10 ng线性化载体,插入DNA与线性化载体摩尔比为10:1时进行SLiCE反应,转化到化转效率为2×105 CFU/μg的感受态细胞没有菌落长出,转化到化转效率为1.32×106 CFU/μg的感受态细胞时产生2个转化子,经PCR验证并非为克隆子。而当转化到化转效率为1.16×107 CFU/μg的感受态细胞时,虽然只产生7个转化子,但经菌落PCR验证重组效率高达100%。表明在本实验中感受态细胞效率达到1.16×107 CFU/μg时,SLiCE便可成功构建出重组质粒。
但是采用Inoue法制备化转效率为1.16×107 CFU/μg的感受态细胞仍较为繁琐。为此我们采取了本实验优化的SLiCE方案:采用线性化载体浓度为50 ng,插入DNA与线性化载体摩尔比为10:1,先进行SLiCE反应,后退火处理。再将SLiCE反应产物转化至CaCl2法制备的化转效率为1.7×106 CFU/μg感受态细胞,也产生2个转化子,且经过PCR验证后均为克隆子。本实验优化的SLiCE方案,即使用CaCl2法制备得到的感受态细胞也能实现无缝克隆。
虽然经过一系列的优化后可以在较低感受态效率的条件下实现SLiCE无缝克隆,但是产生的克隆子过少。我们推测SLiCE克隆中的一些盐,如SLiCE buffer和退火缓冲液都含有一定浓度的盐类,都会一起随插入片段和线性化载体转化,而一些盐往往会降低感受态细胞的转化效率。本研究检测了退火缓冲液对感受态细胞转化效率的影响,感受态细胞转化pUC19质粒的效率为3.6×107 CFU/μg,而在转化中加入退火缓冲液的化转效率降低到6×106 CFU/μg,表明退火缓冲液虽然促进了线性化载体和插入DNA同源区域退火形成连接,但也有可能影响到了转化效率。
为进一步提升转化效率,采用硅藻土纯化的方式对最终的SLiCE反应产物进行纯化,再化转到转化效率为1.7×106 CFU/μg感受态细胞中。虽然相比未经纯化的处理的SLiCE反应产物多产生了一倍的转化子,但PCR验证转化效率仅为50%(图 3)。

|
| M:DL 5000 marker;C:以pUC19质粒为模板进行PCR作为阴性对照;1、2:验证未经纯化的SLiCE催化产物化转到1.7×106 CFU/μg感受态细胞后产生的克隆子;3-6:验证经纯化的SLiCE催化产物化转到1.7×106 CFU/μg感受态细胞后产生的克隆子 图 3 SLiCE反应产物的纯化对克隆的影响 |
Zhang等[7]所建立的SLiCE(Seamless ligation cloning extract)无缝克隆方法在同源臂长度为15-19 bp条件下就能很好的完成质粒构建。Motohashi[13]和Okegawa[14, 17]对这一方法进行了进一步优化,改用E.col JM109进行细胞裂解液的制备,且采用更便宜的非离子表面活性剂来对细胞进行裂解,大大降低了每次反应的成本,使SLiCE成为最便宜的无缝克隆方法之一[14]。但是该无缝克隆技术需要转化效率约为1×108 CFU/μg的感受态细胞,购买高感受态细胞使得整个实验成本居高不下,而自己制备高感受态细胞转化效率往往不稳定。
降低无缝克隆对高感受态细胞的依赖性,需要继续提高无缝克隆的克隆效率。王广珺等[15]通过添加退火缓冲液进行退火处理,将依赖T4聚合酶外切酶活性的无缝克隆的重组效率提高了10倍之多,方法简单且有效。考虑现有的无缝克隆技术及其涉及的相关商品化试剂盒大多也是基于外切酶的应用,如核酸外切酶Ⅲ[21]、T4 DNA聚合酶[22]、T5 DNA聚合酶[6]、λ核酸外切酶[23-24]、Exo Ⅷ[25]等,它们共同的特点是将插入DNA片段和载体两端外切,形成单链的同源重组臂,从而实现无缝克隆。因此,添加退火缓冲液进行退火处理,对这些基于核酸外切酶的无缝克隆技术来说都有提高克隆效率的可能。
最近Nozaki[16]的研究表明大肠杆菌体内具有3'-5'外切酶活性的酶如Xth A,该酶也能促进核酸片段重组,据此我们推测E.col JM109细胞裂解液中也可能含有核酸外切酶。因此,本研究也采用添加退火缓冲液进行退火处理的方式来优化SLiCE技术(图 4)来进一步提高SLiCE技术的克隆效率。

|
| 蓝色区域和黄色区域为SLiCE反应所需的同源区域;红色方框内为加入的优化步骤;采用双酶切法制备线性化载体,酶切后产生非同源序列悬突 图 4 优化的SLiCE流程图 |
在引入变性和复性的步骤之前,我们首先对线性化载体的最佳浓度进行了探索。传统的酶连接多采用20-100 ng线性化载体进行反应。Zhang等[7]认为SLiCE反应中加入10-200 ng的线性化载体和1-10倍线性化载体摩尔浓度的插入DNA的量较为合适。我们的研究发现当参与SLiCE反应的线性化载体的量从10 ng升高到50 ng时,克隆效率提高了近4倍(表 5),但是随着浓度进一步提高,克隆子的数量并没有明显的提高,这可能受限于细胞裂解液中的相关外切酶的酶活。在尽可能减少线性化载体用量且保障克隆效率的原则下,本研究选用50 ng线性化载体进行实验发现,先进行SLiCE反应,再进行退火程序的方式有利于增强SLiCE克隆效率。推测可能原因,先进行SLiCE反应,可通过外切酶作用将插入DNA片段或者载体两端形成单链悬突,此时再进行退火程序,在退火缓冲液和缓慢降温过程的共同作用下,单链DNA(ssDNA)中可能形成的二级结构全部打开,可更准确地使插入DNA片段和载体同源区域的碱基产生氢键互补配对,重组形成新的质粒,进而提高重组效率。而先退火处理,再进行SLiCE反应这种方式,退火程序使线性片段和插入DNA片段的双链都打开,所生成的ssDNA在同源区域也不会形成二级结构,但此时除了线性化载体和插入DNA同源区域产生碱基互补配对外,还有线性化载体的两条ssDNA之间、插入DNA的两条ssDNA之间的竞争性碱基互补配对,这可能导致了重组效率的降低。
即使通过DNA片段纯化,电转化也不能满足SLiCE克隆的要求。电转化相对于化学转化,往往具有更高的转化效率,且制作感受态细胞的过程更为简单。更高的转化效率意味更多的DNA片段或者质粒能进入细胞,产生更多的克隆子,但以往的无缝克隆研究中发现,电转化取得的效果并不理想[6]。有研究者认为,电转化过程中产生大量热量会导致DNA片段形成错误的二级结构[26],也有人认为电转化不同于化转,只会形成少数孔洞,不像化学转化那样保证充足DNA片段转化到每个细胞。我们认为,影响电转化效率最重要的原因是没有去除反应体系中离子,造成了最终转化效率的下降。实验也进一步验证了我们的结果,当SLiCE反应后我们加入了退火缓冲液进行了退火处理,这种SLiCE反应产物直接去实施电转化,没有任何克隆子的产生,而纯化后,产生了761个转化子,虽然最终的重组效率仅为6.25%,但获得克隆子的数目还是不少于化学转化方法的。只是电转化方法产生的假阳性克隆太多,增加了筛选压力,因此不建议在SLiCE克隆中使用电转化。
4 结论本实验对SLiCE反应所需DNA浓度进行优化后,发现当线性化载体量从10 ng升高到50 ng,插入DNA片段与载体的摩尔比为10:1时,SLiCE的克隆效率提高了近4倍。在SLiCE反应之后加入退火缓冲液并进行退火程序,不仅能够提高SLiCE的克隆效率,而且使得SLiCE能利用CaCl2法制备的转化效率仅为1.7×106 CFU/μg的感受态细胞完成重组转化,克服了SLiCE对高感受态细胞的依赖性,进一步降低了SLiCE克隆的成本。
| [1] |
Cohen SN, Chang ACY. Construction of biologically functional bacterial plasmids in vitro[J]. Proc Natl Acad Sci USA, 1973, 70(11): 3240-3244. DOI:10.1073/pnas.70.11.3240 |
| [2] |
Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC[J]. Nat Methods, 2007, 4(3): 251-256. DOI:10.1038/nmeth1010 |
| [3] |
Klock HE, Koesema EJ, Knuth MW, et al. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts[J]. Proteins, 2008, 71(2): 982-994. DOI:10.1002/prot.21786 |
| [4] |
Gibson DG, Young L, Chuang RY, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases[J]. Nat Methods, 2009, 6(5): 343-345. DOI:10.1038/nmeth.1318 |
| [5] |
Quan J, Tian J. Circular polymerase extension cloning of complex gene libraries and pathways[J]. PLoS One, 2009, 4(7): e6441. DOI:10.1371/journal.pone.0006441 |
| [6] |
Xia Y, Li K, Li J, et al. T5 exonuclease-dependent assembly offers a low-cost method for efficient cloning and site-directed mutagenesis[J]. Nucleic Acids Res, 2019, 47(3): e15. DOI:10.1093/nar/gky1169 |
| [7] |
Zhang Y, Werling U, Edelmann W. SLiCE:a novel bacterial cell extract-based DNA cloning method[J]. Nucleic Acids Res, 2012, 40(8): e55. DOI:10.1093/nar/gkr1288 |
| [8] |
Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda[J]. Microbiol Mol Biol Rev, 1999, 63(4): 751-813. DOI:10.1128/MMBR.63.4.751-813.1999 |
| [9] |
Muyrers JP, Zhang Y, Buchholz F, et al. RecE/RecT and Reda/Redb initiate double-stranded break repair by specifically interacting with their respective partners[J]. Genes Dev, 2000, 6(14): 1971-1982. |
| [10] |
Lovett ST, Hurley RL, Jr VAS. Crossing over between regions of limited homology in Escherichia coli :RecA-dependent and RecA-independent pathways[J]. Genetics, 2000, 160: 851-859. |
| [11] |
Dutra BE, Sutera VA, Lovett ST. RecA-independent recombination is efficient but limited by exonucleases[J]. Proc Natl Acad Sci USA, 2007, 104(1): 216-221. DOI:10.1073/pnas.0608293104 |
| [12] |
Persky NS, Lovett ST. Mechanisms of recombination:lessons from E. coli[J]. Crit Rev Biochem Mol Biol, 2008, 43(6): 347-370. DOI:10.1080/10409230802485358 |
| [13] |
Motohashi K. A simple and efficient seamless DNA cloning method using SLiCE from Escherichia coli laboratory strains and its application to SLiP site-directed mutagenesis[J]. BMC Biotechnol, 2015, 15(47): 1-9. |
| [14] |
Okegawa Y, Motohashi K. A simple and ultra-low cost homemade seamless ligation cloning extract(SLiCE)as an alternative to a commercially available seamless DNA cloning kit[J]. Biochem Biophys Rep, 2015, 4: 148-151. |
| [15] |
王广珺, 何川, 张义正. 不依赖基因序列和连接反应克隆(SLIC)方法的改进[J]. 四川大学学报, 2010, 7, 4(47): 893-898. |
| [16] |
Nozaki S, Niki H. Exonuclease Ⅲ(XthA)enforces in vivo DNA cloning of Escherichia coli to create cohesive ends[J]. J Bacteriol, 2018, 201(5): pii: e00660-18. |
| [17] |
Okegawa Y, Motohashi K. Evaluation of seamless ligation cloning extract preparation methods from an Escherichia coli laboratory strain[J]. Anal Biochem, 2015, 6(486): 51-53. |
| [18] |
余宏傲, 张岚岚, 徐昌杰. 一种有效纯化低拷贝质粒DNA的改良硅藻土法[J]. 细胞生物学杂志, 2005, 27: 679-683. |
| [19] |
Inoue H, Nojima H, 0kayama H. High efficiency transformation of Escherichia coli with plasmids[J]. Gene, 1990, 96: 23-28. DOI:10.1016/0378-1119(90)90336-P |
| [20] |
Li JF, Li L, Sheen J. Protocol:a rapid and economical procedure for purification of plasmid or plant DNA with diverse applications in plant biology[J]. Plant Methods, 2010, 6(1): 1-8. DOI:10.1186/1746-4811-6-1 |
| [21] |
Hsiao KC. Exonuclease Ⅲ induced ligase-free directional subcloning of PCR products[J]. Nucleic Acids Res, 1993, 21(23): 351-359. |
| [22] |
Islam MN, Lee KW, Yim HS, et al. Optimizing T4 DNA polymerase conditions enhances the efficiency of one-step sequence- and ligation-independent cloning[J]. Biotechniques, 2017, 63(3): 125-130. |
| [23] |
Kovall R, Matthews BW. Toroidal structure of lambda-exonuclease[J]. Science, 1997, 277(5333): 1824-1827. DOI:10.1126/science.277.5333.1824 |
| [24] |
Wu T, Yang Y, Chen W, et al. Noncanonical substrate preference of lambda exonuclease for 5'-nonphosphate-ended dsDNA and a mismatch-induced acceleration effect on the enzymatic reaction[J]. Nucleic Acids Res, 2018, 46(6): 3119-3129. DOI:10.1093/nar/gky154 |
| [25] |
朱燕, 韩小韦, 牛毅男, 等. 核酸外切酶Ⅷ截短体的表达及其在体外DNA重组反应中的应用[J]. 生物工程学报, 2019, 35(6): 1-10. |
| [26] |
Benoit RM, Ostermeier C, Geiser M, et al. Seamless insert-plasmid assembly at high efficiency and low cost[J]. PLoS One, 2016, 11(4): e0153158. DOI:10.1371/journal.pone.0153158 |