




2. 中国农业科学院兰州畜牧与兽药研究所,兰州 730050
2. Lanzhou Institute of Husbandry and Pharmaceutical Science, Chinese Academy of Agricultural Sciences, Lanzhou 730050
土壤盐渍化是全球性的生态问题, 不仅导致生态环境恶化, 而且也是植物生长的主要非生物胁迫因素[1]。我国盐渍化危害严重, 各类盐碱地面积约9 913万hm2[2]。如何高效利用这些盐碱化土地已成为一个重要的课题和研究方向[3]。利用盐生植物进行盐碱地生物改良是盐碱地恢复和改良的重要途径, 而选择适宜的种植材料是实施盐碱地生物恢复和改良的关键。目前, 适宜于盐碱地改良的植物种类比较有限, 不同种源的同一植物种耐盐性差异也较大, 因此, 有必要通过选育和培育抗盐品种来提高植物的抗盐性[4]。为此, 在分子生物学层面上阐明植物的耐盐机制, 获得植物的耐盐相关基因, 通过现代生物技术实现耐盐基因转化, 并以此提高植物耐盐性已成为目前耐盐植物研究的热点[5-6]。
黑果枸杞(Lycium ruthenicum Murr.)为茄科(Solanceae)枸杞属(Lycium L.)多年生灌木, 分布于新疆、青海、甘肃、宁夏和内蒙古等地[7]。常以灌丛状生于盐碱荒地、盐化沙地、盐湖岸边、道路两侧集水区等各种盐渍化土壤或荒漠环境, 是我国荒漠区特有的耐盐抗旱同时具有很高经济价值及营养价值的野生植物[8-9]。黑果枸杞具有抗旱、抗寒、耐盐碱、根蘖性强、耐土壤贫瘠及营养价值高等诸多优点, 是盐碱地治理的先锋树种, 具有广泛的经济前景和开发潜力[10-11]。目前有关黑果枸杞抗盐性研究主要集中在生理生化和解剖结构等方面[12-14], 对其分子水平的研究报道则相对较少[15-17]。随着高通量测序技术的快速发展, 转录组测序成为研究基因差异表达的重要手段[18-20]。本研究利用Illumina测序平台, 进行不同浓度盐胁迫处理下的黑果枸杞根和叶的转录组测序, 筛选盐胁迫下叶和根中的差异表达基因, 旨为揭示黑果枸杞响应盐胁迫的分子机制提供基础资料。
1 材料与方法 1.1 材料以甘肃农业大学林学院实验室保存的黑果枸杞组培苗为材料。
1.2 方法 1.2.1 NaCl处理2018年10月, 选取生长健壮、长势一致的黑果枸杞组培苗, 开盖炼苗5 d后, 清洗根部残留的培养基, 转移到塑料盒(口径10 cm, 高10 cm, 带有通气装置)。塑料盒中装有等量含有不同浓度NaCl的1/2 Hoagland营养液进行组培苗盐胁迫处理, 每个处理1株, 3次重复。1/2 Hoagland营养液中添加的NaCl浓度为:0(CK)mmol/L、50 mmol/L和250 mmol/L。上述处理分别在胁迫0、1和12 h时取根和叶(含嫩茎)各0.1 g用于转录组和基因表达谱的分析。
1.2.2 总RNA的提取、文库构建和转录组测序用mirVanaTM miRNA ISOlation Kit(Ambion-1561)试剂盒提取和纯化黑果枸杞叶片和根的总RNA。分别用Nanodrop 2000与Agilent2100 Bioanalyzer进行总RNA质量和纯度检测。分别取NaCl胁迫处理组和对照组的黑果枸杞根和叶样品的RNA, 送样至上海欧易生物医学科技有限公司进行转录组测序。
1.2.3 Unigene功能注释和差异表达基因分析使用blastx[21], 取阈值e < 1e-5, 将Unigene分别注释到NR、KOG和Swissprot数据库。基于Swissprot结果, 将Swissprot ID映射到GO term, 获得Unigene的GO注释, 最后将Unigene比对到KEGG[22]数据库获得通路信息。Unigene的FPKM[21]和count使用bowtie2[22]和eXpress[23]软件分析得到。通过eXpress软件获取落到各个样本中Unigene的reads数目, 使用DESeq[23]R package的estimateSizeFactors函数对数据进行标准化, 并使用nbinomTest函数计算差异比较的pvalue和foldchange值。挑选出P < 0.05, 差异倍数大于2的差异Unigene, 并进行差异Unigene的GO和KEGG[24]富集分析, 以判定差异Unigene主要影响的生物学功能或通路。利用HMMER[24]软件比对Pfam数据库来进行Unigene的功能分析。
2 结果 2.1 总RNA质量检测和组装结果分析经检测, 样品总RNA的A260/A280均大于1.8, 28S/18S ≥ 0.7, RIN ≥ 7, 表明所提RNA可满足后继实验分析需求。
黑果枸杞幼苗在不同浓度NaCl胁迫和不同取样时间下叶和根共30个样本, 通过转录组测序共获得222.49 G的原始数据, 各样本的Q30碱基比均在95%以上, GC含量均在43%以上(表 1)。说明测序结果准确度较高, 可用于后续分析。利用Trinity软件对测序所得数据进行合并组装, 共获得86 037个Unigene, 其中长度在1 kb以上的Unigene有31 070条, 这些Unigene可作为后续实验研究的重点对象; Unigene平均长度为1 097.27 bp(表 2)。


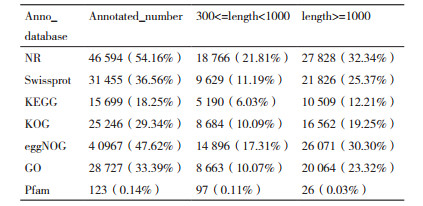
由表 3可知, 盐胁迫下黑果枸杞根和叶的86 037个Unigene中有47 108个Unigene在不同数据库中的得到了注释, 占总Unigened的54.76%, 还有38 929个Unigene在这些数据库中没有得到注释。其中注释到NR数据库的Unigene最多, 达到46 594个, 占54.16%;注释到Pfm数据库的Unigene最少, 仅有123个, 占0.14%;共有16个Unigene在所有数据库中都得以注释。
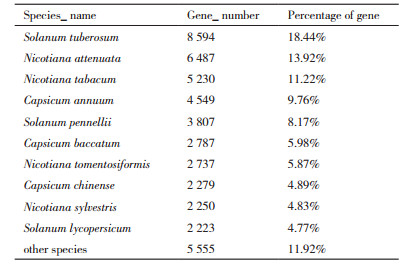
由表 4可知, 黑果枸杞转录组所得Unigene在NR数据库注释中与马铃薯(Solanum tuberosum)同源序列最多, 为8 594个, 占注释Unigene的18.48%, 番茄(Solanum lycopersicum)同原序列最少, 为2 223个, 占注释Unigened 4.78%, 其他物种的5 555, 占注释Unigene的11.95%。
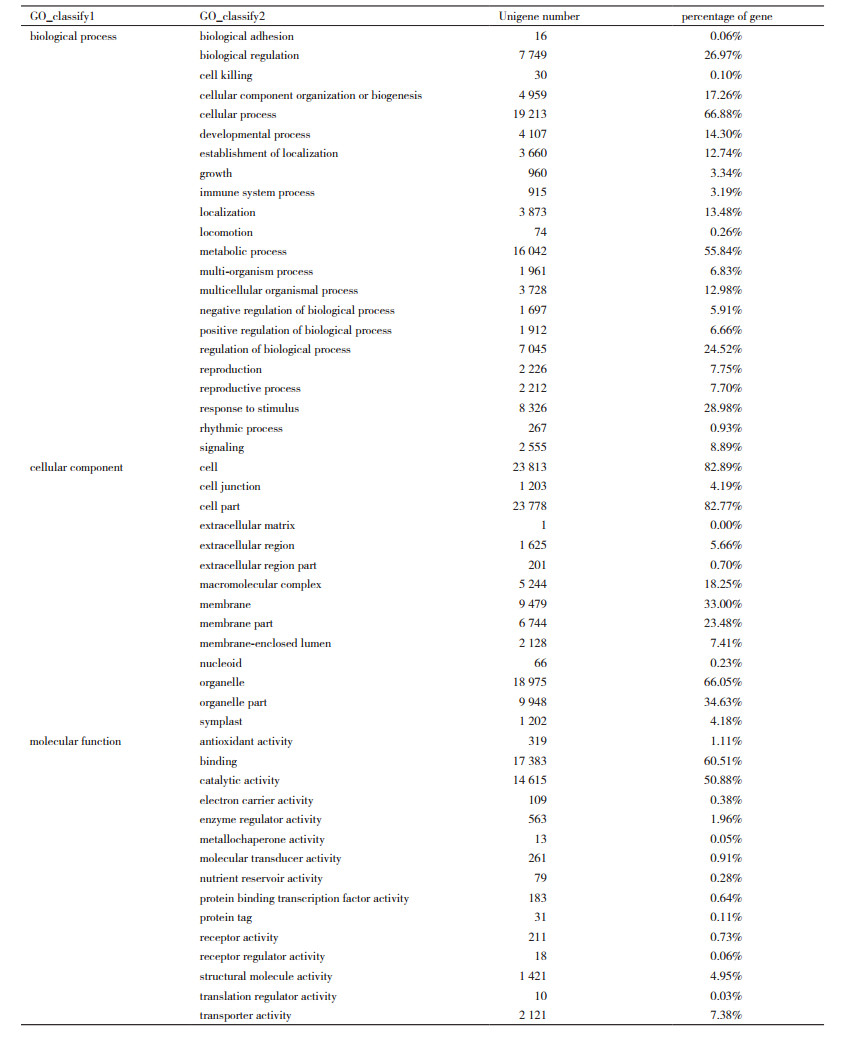
对获得相应GO条目的Unigenes进行统计分析(表 5), 共有28 727个Unigenes获得235 271个GO注释, 平均每条8.19个。获得的GO数据库注释的Unigene可分为细胞组分(Cellular component)、分子功能(Molecular function)和生物过程(Biological process)3个GO类别的51个小组。细胞组分涉及104 407个GO条目, 分为14个功能组, 其中细胞(Cell, 23 813个)、细胞部分(Cell part, 23 778个)、细胞器(Organelle, 18 975)和细胞膜(Membrane, 9 479个)涉及的Unigene较多; 分子功能涉及37 337个GO条目, 分为15个功能组, 其中催化活性(Catalytic activity, 14 615个)和结合(Binding, 17 383个)含Unigene较多; 93 527个GO条目归属于生物过程, 分为22个功能组, 其中代谢过程(Metabolic process, 16 042个)、细胞进程(Cellular process, 19 213个)和刺激响应(Response to stimulus, 8 326个)涉及的Unigene较多。
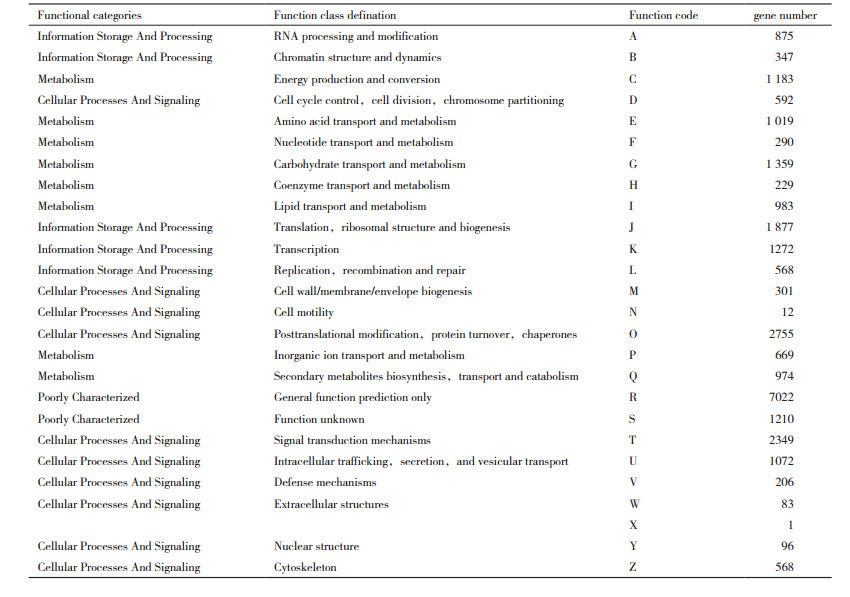
为了进一步分析黑果枸杞根和叶转录组Unigene的功能, 进行了KOG功能分类分析, 共有25 246个Unigenes获得27 912个KOG注释, 平均每条1.1个。分类结果如表 6所示, 共获得25个不同的功能分类。一般功能预测(General function prediction only)的Unigene为7 022条, 是最大的功能类群; 其次是翻译后修饰、蛋白质翻转和分子伴侣(Post translation almodification, protein turnover, chaperones)2 755条, 信号转导类机制(Signal transduction mechanisms)次之, 有2 349条, 最少的是细胞活性(Cell motility)功能类别, 仅12条。
注释到KEGG的15 699个Unigenes获得15 844个KEGG注释。参与的代谢通路可归为4大类别、24个子类。由表 7可知, 4种代谢通路大类中, 与代谢(Metabolism)相关的通路获得8 143个注释, 遗传信息处理(Genetic information processing)3 893个, 细胞过程(Cellular processes)与环境信息处理(Environmental information processing)分别是1 955个和1 853个。进一步细分为24个子类代谢途径, 其中, 翻译(Translation)获得注释最多, 为1 983个; 其次为信号传导(Signal transduction)和碳水化合物代谢(Carbohydrate metabolism), 分别为1 777个和1 512个。
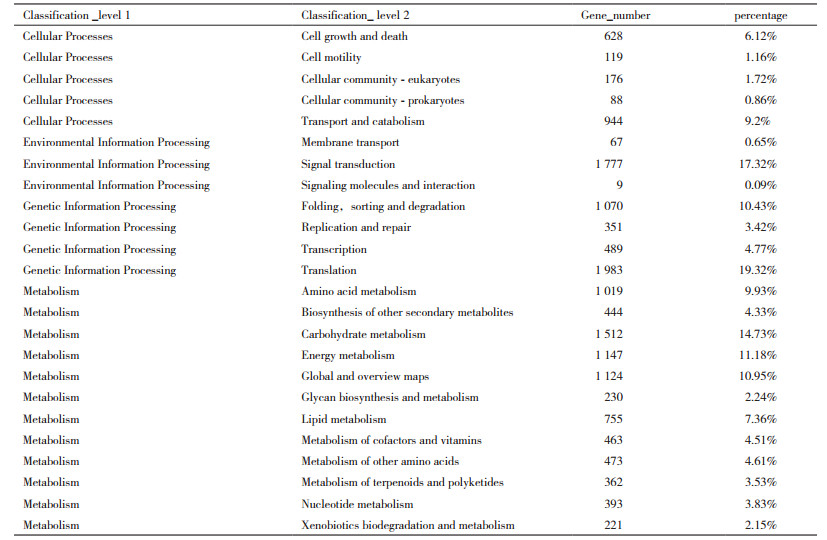
共有21 215个Unigenes归入211条代谢通路, 靠前的有14个代谢通路(表 8)。核糖体(Ribosome)代谢途径注释到的Unigenes数量最多, 有1 241个Unigenes; 其次为碳代谢(Carbon metabolism)723、氨基酸的生物合成(Biosynthesis of amino acids)546和内质网蛋白处理(Protein processing in endoplasmic reticulum)490等途径。

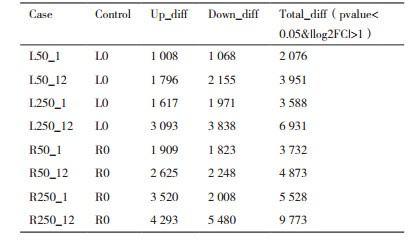
由表 9可以看出, 黑果枸杞在不同浓度NaCl和不同处理时间下与CK相比, 叶片和根随着NaCl浓度增大和处理时间延长, 上调基因和下调基因数呈增加趋势; 叶片上调基因数(7 514)小于下调基因数(9 032), 而根的上调基因数(12 347)大于下调基因数(11 559)。
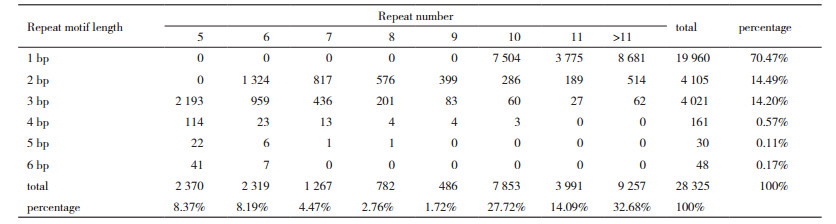
利用软件MISA对黑果枸杞盐胁迫下转录组测序所获得的Unigene进行SSR预测, 结果见表 10。由表 10可知, 共有28 325个SSR位点, 单核苷酸SSR最多, 为19 960, 占70.47%;5核苷酸SSR最少, 为0.11%。重复单元重复出现的次数大于11次以上最多, 为9 257, 占32.68%;重复单元重复出现7次的最少, 为486, 占1.72%。
对于未完成全基因组测序的物种来说, 采用转录组测序技术可获得大量的转录本信息, 这可为植物基因表达的综合分析提供合理可靠的数据资源[25]。因此, 对黑果枸杞在盐胁下的转录组信息进行系统分析, 为全面了解黑果枸杞抗盐分子机制和挖掘新的抗盐基因提供基础资料。本研究在没有参考基因组测序情况下, 对不同浓度NaCl溶液胁迫不同时间时的黑果枸杞组培苗的叶片和根进行转录组测序, 共获得86 037条Unigenes。将这些Unigene在NR、Swissprot、KEGG、KOG、eggNOG、GO及Pfam数据库中进行注释, 共有47 108个Unigene得到了注释, 占总Unigened的54.76%, 还有38 929个Unigene没有得到注释。这一结果在目前已测序的许多植物当中都存在[26], 即并不是所有组装得到的Unigene都能在已知的有关基因数据库得到注释。这是由于大量非模式植物的基因组测序工作没有进行, 缺少相关基础研究数据, 需要对这些没有注释的基因进一步分析研究, 来确定这些基因在植物生长发育过程中的作用, 从而丰富基因数据库。
植物在受到盐胁迫时会产生大量转导信号, 有离子信号转导、渗透信号转导、解毒类信号转导等; 同时植物体内蔗糖和淀粉的分解代谢过程加强, 分解的糖类不仅为植物生长发育提供能量, 而且分解产生的小分子单糖提高了细胞中可溶性糖的含量和细胞的渗透势, 以抵抗高浓度盐离子带来的渗透胁迫[27]。本研究在KEGG代谢途径注释中翻译(Translation)获得注释最多, 其次为信号传导(Signal transduction)和碳水化合物代谢(Carbohydrate metabolism)。这是因为当黑果枸杞受到盐胁迫时, 自身的碳水化合物进行代谢来产生足够的能量使植物能够进行各种生理反应, 植物需要合成大量的酶来催化体内的各种生理生化反应, 同时将受到的刺激信号进行传导, 以便植物能产生应激反应来应对不良环境造成的影响。这一结果与滨柃[28]、辽宁碱蓬[29]、海滨锦葵[30]等植物在盐胁迫下转录组分析结果基本一致, 这也说明不同植物在盐胁迫有相似的抗盐机制。
在DEGs分析中, 叶的上调基因数(7 514)小于下调基因数(9 032), 而根的上调基因数(12 347)大于下调基因数(11 559)。这是由于在盐胁迫下, 植物根系最早感受逆境胁迫信号, 并产生相应的生理反应, 继而影响地上部生长[31]。张倩倩[32]在对中国石竹研究过程也发现, 在盐胁迫过程中根中的DEGs比叶中高。这也说明当黑果枸杞受到盐胁迫时根部就可能会诱导更多与抗盐有关基因表达, 使得黑果枸杞能够在盐胁迫下正常生长, 而叶片中可能与抗盐关系不大的基因被抑制, 使得黑果枸杞与抗盐关系不明显的性状得以抑制, 把更多能量用于与抗盐有关的性状生长。需要进一步的对这些差异基因进行分析, 筛选出与黑果枸杞抗盐有关的基因, 从分子生物学角度来解释黑果枸杞抗盐机制。
SSR是以特异引物PCR为基础的分子标记技术[33], 因其多态性高、共显性遗传、覆盖面广等优点[34], 广泛运用于遗传图谱构建、目标基因的标定、遗传多样性分析和分子标记辅助育种等方面[35]。已对许多物种进行了基于转录组SSR信息的分析研究, 如马尾松(Pinus massoniana)[36]、蓝靛果忍冬(Lonicera caerulea)[37]、山桐子(Idesia polycarpa)[38]等。这些研究结果表明转录组中具有丰富的SSR位点信息, 单核苷酸、二核苷酸和三核苷酸为优势重复类型在不同物种中不同。本研究利用MISA对黑果枸杞盐胁迫下转录组测序所获得的Unigene进行SSR预测, 共发现28 325个SSR位点, 优势重复基序为单核苷酸, 占总SSR位点的70.47%, 这一研究结果与尹跃等[39]的结果一致, 即黑果枸杞转录组的SSR主要类型为单核苷酸重复基序(74.33%)。
4 结论本研究利用Illumina HiSeq X Ten测序仪对不同浓度NaCl溶液胁迫下的黑果枸杞转录组进行测序, 转录组测序共产生222.49 Gb原始数据, 拼接出Unigenes 86 037条, 注释到7大功能数据库(GO、KEGG、KOG、NR、Pfam、Swiss-Prot和egg NOG)上的Unigenes总数为46 594个, 还有38 929个Unigenes在这些数据库中没有得到注释。通过GO分类和KEGG Pathway富集性分析, 将分别归于51个GO类别和211条代谢途径。差异表达基因分析显示, 黑果枸杞叶片叶片中的上调基因数(7 514)小于下调基因数(9 032), 而根中的上调基因数(12 347)大于下调基因数(11 559)。在黑果枸杞盐胁迫下转录组中发现28 325个SSR位点, 最多的为单核苷酸SSR, 占70.47%, 重复单元重复出现的次数大于11次以上最多, 占32.68%。
| [1] |
Oyiga BC, Ogbonnaya FC, Sharma RC, et al. Genetic and transcriptional variations in NRAMP-2 and OPAQUE1 genes are associated with salt stress response in wheat[J]. Theoretical and Applied Genetics, 2019, 132(2): 323-346. DOI:10.1007/s00122-018-3220-5 |
| [2] |
郭世乾, 崔增团, 傅亲民. 甘肃省盐碱地现状及治理思路与建议[J]. 中国农业资源与区划, 2013, 34(4): 75-79. |
| [3] |
赵振勇, 张科, 王雷, 等. 盐生植物对重盐渍土脱盐效果[J]. 中国沙漠, 2013, 33(5): 1420-1425. |
| [4] |
Zhao K, Song J, Feng G, et al. Species, types, distribution, and economic potential of halophytes in China[J]. Plant and Soil, 2011, 342(1-2): 495-509. DOI:10.1007/s11104-010-0470-7 |
| [5] |
Štefanić PP, Koffler T, Adler G, et al. Chloroplasts of salt-grown Arabidopsis seedlings are impaired in structure, genome copy number and transcript levels[J]. PLoS One, 2013, 8(12): e82548. DOI:10.1371/journal.pone.0082548 |
| [6] |
Lee SY, Seok HY, Tarte VN, et al. The Arabidopsis chloroplast protein S-RBP11 is involved in oxidative and salt stress responses[J]. Plant Cell Reports, 2014, 33(6): 837-847. DOI:10.1007/s00299-013-1560-9 |
| [7] |
中国植物志编辑委员会. 中国植物志(第67卷第一册)[M]. 北京: 中国科学出版社, 1999.
|
| [8] |
Wang H, Li J, Tao W, et al. Lycium ruthenicum studies :Molecular biology, phytochemistry and pharmacology[J]. Food Chemistry, 2018, 240: 759-766. DOI:10.1016/j.foodchem.2017.08.026 |
| [9] |
Lv X, Wang C, Cheng Y, et al. Isolation and structural characterization of a polysaccharide LRP4-A from Lycium ruthenicum Murr[J]. Carbohydrate Research, 2013, 365: 20-25. DOI:10.1016/j.carres.2012.10.013 |
| [10] |
Li YH, Zou XB, Shen TT, et al. Determination of geographical origin and anthocyanin content of black goji berry(Lycium ruthenicum Murr.)using near-infrared spectroscopy and chemometrics[J]. Food Analytical Methods, 2017, 10(4): 1034-1044. DOI:10.1007/s12161-016-0666-4 |
| [11] |
刘丽萍, 张东智, 张冲, 等. 黑果枸杞抗逆性及栽培育种研究进展[J]. 生物技术通报, 2016, 32(10): 118-127. |
| [12] |
马彦军, 许晶晶, 韩谨如, 等. 3个种群黑果枸杞叶片解剖结构的耐盐性分析[J]. 干旱区资源与环境, 2018, 32(4): 100-105. |
| [13] |
张荣梅, 马彦军. NaCl胁迫对黑果枸杞叶片生理指标的影响[J]. 甘肃农业大学学报, 2018, 52(4): 110-157. |
| [14] |
武燕, 尹建军, 李善家. 黑河下游荒漠植物黑果枸杞叶片性状特征及其盐分响应[J]. 生态学杂志, 2017, 36(5): 1277-1284. |
| [15] |
Chen JH, Zhang DZ, Zhang C, et al. Physiological characterization, transcriptomic profiling, and microsatellite marker mining of Lycium ruthenicum[J]. Journal of Zhejiang University-Science (Biomedicine & Biotechnology), 2017, 18(11): 1002-1021. |
| [16] |
严莉, 王翠平, 陈建伟, 等. 基于转录组信息的黑果枸杞MYB转录因子家族分析[J]. 中国农业科学, 2017, 50(20): 3991-4002. |
| [17] |
王翠平, 陈建伟, 严莉, 等. 黑果枸杞R1-MYB转录因子基因的克隆及表达分析[J]. 中草药, 2018, 49(1): 203-210. |
| [18] |
Gruber MY, Xia J, Yu M, et al. Transcript analysis in two alfalfa salt tolerance selected breeding populations relative to a nontolerant population[J]. Genome, 2016, 60(2): 104-127. |
| [19] |
Mansouri M, Naghavi MR, Alizadeh H, et al. Transcriptomic analysis of Aegilops tauschii during long-term salinity stress[J]. Functional & Integrative Genomics, 2019, 19(1): 13-28. |
| [20] |
Postnikova OA, Shao J, Nemchinov LG. Analysis of the alfalfa root transcriptome in response to salinity stress[J]. Plant and Cell Physiology, 2013, 54(7): 1041-1055. DOI:10.1093/pcp/pct056 |
| [21] |
Bolger AM, Lohse M, Usadel B. Trimmomatic :a flexible trimmer for Illumina sequence data[J]. Bioinformatics, 2014, 30(15): 2114-2120. DOI:10.1093/bioinformatics/btu170 |
| [22] |
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2[J]. Nature Methods, 2012, 9(4): 357-359. DOI:10.1038/nmeth.1923 |
| [23] |
Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation[J]. Nature Biotechnology, 2010, 28(5): 511-515. DOI:10.1038/nbt.1621 |
| [24] |
Grabherr MG, Haas BJ, Yassour M, et al. Trinity :reconstructing a full-length transcriptome without a genome from RNA-Seq data[J]. Nature Biotechnology, 2011, 29(7): 644-652. DOI:10.1038/nbt.1883 |
| [25] |
贾新平, 孙晓波, 邓衍明, 等. 鸟巢蕨转录组高通量测序及分析[J]. 园艺学报, 2014, 41(11): 2329-2341. |
| [26] |
张少平, 洪建基, 邱珊莲, 等. 紫背天葵高通量转录组测序分析[J]. 园艺学报, 2016, 43(5): 935-946. |
| [27] |
Stitt M, Sulpice R, Keurentjes J. Metabolic networks :how to identify key components in the regulation of metabolism and growth[J]. Plant Physiology, 2010, 152(2): 428-444. DOI:10.1104/pp.109.150821 |
| [28] |
赵路遥.盐胁迫下滨柃(Eurya emarginata)幼苗的生理响应和转录组分析[D].舟山: 浙江海洋大学, 2017. http://cdmd.cnki.com.cn/Article/CDMD-10340-1017153630.htm
|
| [29] |
曹盈.盐生植物辽宁碱蓬(Suaeda liaotungensis K.)转录组测序及分析[D].大连: 辽宁师范大学, 2013. http://cdmd.cnki.com.cn/Article/CDMD-10165-1014139177.htm
|
| [30] |
汤晓丽.海滨锦葵盐胁迫转录组分析和抗盐基因研究[D].北京: 中国科学院大学, 2016. http://cdmd.cnki.com.cn/Article/CDMD-80180-1016904143.htm
|
| [31] |
马进, 郑刚. 利用转录组测序技术鉴定紫花苜蓿根系盐胁迫应答基因[J]. 核农学报, 2016, 30(8): 1470-1479. |
| [32] |
张倩倩.短期盐胁迫下中国石竹幼苗响应的转录组测序、组装和分析[D].呼和浩特: 内蒙古农业大学, 2017. http://cdmd.cnki.com.cn/Article/CDMD-10129-1017212518.htm
|
| [33] |
Powell W, Morgante M, Andre C, et al. The comparison of RFLP, RAPD, AFLP and SSR(microsatellite)markers for germplasm analysis[J]. Molecular Breeding, 1996, 2(3): 225-238. DOI:10.1007/BF00564200 |
| [34] |
Song QJ, Shi JR, Singh S, et al. Development and mapping of microsatellite(SSR)markers in wheat[J]. Theoretical and Applied Genetics, 2005, 110(3): 550-560. DOI:10.1007/s00122-004-1871-x |
| [35] |
Eujayl I, Sorrells ME, Baum M, et al. Isolation of EST-derived microsatellite markers for genotyping the A and B genomes of wheat[J]. Theoretical and Applied Genetics, 2002, 104(2-3): 399-407. |
| [36] |
梅利那, 范付华, 崔博文, 等. 基于马尾松转录组的SSR分子标记开发及种质鉴定[J]. 农业生物技术学报, 2017, 25(6): 991-1002. |
| [37] |
张庆田, 李晓艳, 杨义明, 等. 蓝靛果忍冬转录组SSR信息分析及其分子标记开发[J]. 园艺学报, 2016, 43(3): 557-563. |
| [38] |
李娜, 姚民, 梅兰菊, 等. 基于山桐子转录组序列的SSR分子标记开发[J]. 应用与环境生物学报, 2017, 23(5): 952-958. |
| [39] |
尹跃, 安巍, 赵建华, 等. 黑果枸杞转录组SSR信息分析及分子标记开发[J]. 浙江农林大学学报, 2019, 36(2): 422-428. |