




近年来,随着DNA高通量测序技术的快速发展和测序成本的大幅度降低,基于DNA测序技术的转录组学研究已经广泛应用生物学研究之中。利用转录组技术挖掘功能基因是目前最主要的研究方向,是基因功能及结构研究的基础和出发点。转录组测序技术首先被应用于真核生物的基因表达研究,由于原核生物RNA并不生成mRNA,因而其转录组研究起步较晚,但随着RNA测序技术的突破,近年来,原核生物转录组研究发展很快,特别是在致病细菌的致病机理的研究,细菌基因表达对环境胁迫的响应机制,细菌降解环境污染物的分子机制等方面的研究,受到更为广泛地关注,取得了许多重要的研究成果。
1 转录组技术概述转录组的概念是由Charlles Auffary于1996年提出[1],并于1997年首次在文章中被使用。转录组是某一个生物个体、组织或细胞在某一特定的时间或特定环境条件下表达的完整的或近乎完整的转录本的总和。研究转录组对于了解基因组功能及其代谢机理至关重要[2]。目前,常规的转录组测序分析流程可分为实验设计与上机测序。实验设计包括样品的采集和保存、总RNA的提取、RNA质检、目的RNA的富集、cDNA的合成及文库构建。测序后的数据分析包括数据预处理、序列分析与转录本识别、转录本定量与功能分析、差异基因筛选、差异基因功能和代谢通路富集分析、差异基因表达分析以及差异基因功能注释等。
2 原核生物转录组研究现状原核生物暴露于各种环境之中,营养限制、温度、pH、氧气、压力、有机溶剂、重金属、抗生素及其他微生物的存在与关系等,直接地作用于或影响原核生物。原核生物通过调节相应的基因表达以应对环境的变化与环境压力。因此,转录组分析能够准确有效地揭示原核生物响应环境的机制。
2.1 细菌降解污染物的转录组研究2.1.1细菌响应重金属的转录组研究
重金属污染是一个重大的环境问题,利用微生物去除或减少土壤或水体中重金属污染,由于其低成本和生态友好的性质,目前成为研究的热点。微生物可以通过氧化还原作用改变特定金属价态,降低重金属的生物可利用性或潜在的毒性[3]。转录组技术揭示了细菌作用于重金属的机制(表 1)。在Ag(Ⅰ)、Pd(Ⅱ)和Se(Ⅳ)胁迫下,Pantoea sp. IMH显示了对所有金属相同的抗性机制,即多重抗应激蛋白BhsA和谷胱甘肽的解毒代谢。阳离子金属(Ag+和Pd2+)的抗性机制主要是锌转运蛋白和硫酸盐代谢,而草酸转运蛋白和砷的抗性机制则具体参与了对阴离子(SeO32-)的抗性和还原。此外,推测Ag(Ⅰ)可通过葡萄糖作用转化为AgNPs,细胞色素CpxP参与Pd(Ⅱ)的还原。研究结果为Ag(Ⅰ)、Pd(Ⅱ)和Se(Ⅳ)的耐药和降低机制提供了新的线索[4]。
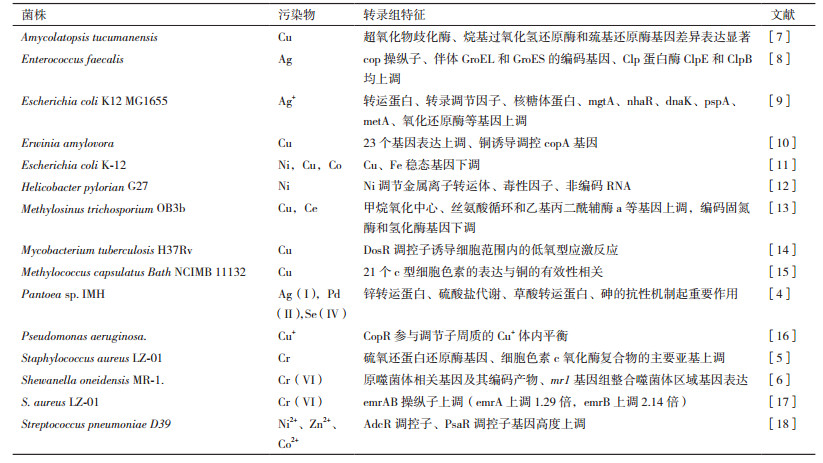
Cr(Ⅵ)具有高可溶性和毒性,长期接触会导致突变和致癌。Cr的另一种最稳定、最常见的形式Cr(Ⅲ)被认为毒性小于Cr(Ⅵ),然而Cr(Ⅲ)会引起DNA损伤,抑制细菌拓扑异构酶DNA弛豫激活。Staphylococcus aureus LZ-01具有将Cr(Ⅵ)有氧还原为Cr(Ⅲ)的能力。转录组测序分析表明,编码硫氧还蛋白还原酶的基因和细胞色素c氧化酶复合物的主要亚基在Cr(Ⅵ)处理后均上调。29个abc型金属/多药转运体和外排泵被上调,表明它们通过将Cr+泵出细胞来参与Cr(Ⅵ)抗性。26个DNA修复基因的上调说明Cr(Ⅵ)对DNA具有毒性,这些DNA保护蛋白需要对Cr(Ⅵ)应激作出反应[5]。Cr(Ⅵ)处理Shewanella oneidensis MR-1 24 h后培养上清液中未检测到残留Cr(Ⅵ),说明细胞完全吸收和/或还原了这种金属。转录组和蛋白组分析发现,基因和蛋白表达谱的差异包括编码铁结合和转运功能的基因的下调或低水平诱导(2-4倍)[6]。
2.1.2 芳香族化合物降解菌的转录组研究了解降解的分子机制对制定有效的生物修复策略至关重要。近年来,有机污染物降解细菌的转录组学研究日益增多(表 2)。多环芳烃在人类影响的环境中普遍存在[19],这可能与代表性污染源(如高温燃烧、交通排放、生物质燃烧)的大量释放[20]和各种分散过程有关。环境中的多环芳烃有很大一部分可以被微生物去除,多环芳烃的微生物降解已经成为一种重要的修复技术[21-22]。转录组和蛋白质组学分析揭示了Bacillus subtilis对芳香族化合物的代谢机制,发现yfiDE(catDE)操纵子编码一种儿茶酚2,3-双加氧酶,这种酶是由儿茶酚强烈诱导的。yodED(mhqED)、ydfNOP(mhqNOP)操纵子和ykcA(mhqA)编码对苯二酚特异性的外二醇加双氧酶[21]。Rhodococcus jostii RHA1能降解多氯联苯(PCB),转录组分析发现该菌株具有6个新ORFs参与促进PCB代谢,其中Ro10225蛋白对RHA1中PCB/联苯双氧合酶复合物的形成至关重要[22]。Corynebacterium jeikeium K411降解香草醇的转录组分析发现,4-羟基-3-甲氧苄醇可导致95个基因的差异表达,其中86个基因上调,耐药决定因子cmx和预测的毒力因子sapA和sapD的表达水平显著提高。Mycobacterium sp. A1-PYR降解不同多环芳烃的转录组分析表明,3种多环芳烃降解酶在二元底物中的表达水平明显高于仅在吡咯底物中的表达水平[23]。这些酶构成了一个完整的酶系统,实现了PYR的所有转化步骤,其大部分编码基因在A1-PYR菌株的基因组中形成了一个新的基因级联,为微生物降解难降解的多环芳烃与易降解的多环芳烃共代谢提供了分子视角。氯脲-乙基是一种典型的长期残留磺脲类除草剂,长期滞留对轮作作物危害很大。微生物降解被认为是最可接受的去除方法,但其降解机理尚不清楚。R. erythropolis D310-1是氯霉素-乙基高效的降解菌,转录组结果表明,菌株D310-1在氯霉素-乙基降解过程中上调了500个基因。主要KEGG代谢途径为甲苯降解和氨基苯甲酸酯降解[24]。

烃类属于致癌物质和神经毒性有机污染物[25]。已经发现超过了200种的微生物可以降解石油烃[26],79个种属的细菌可以利用烷烃作为唯一的碳源和能源[27]。将Acinetobacter venetianus RAG-1分别培养在十二烷、十二烷醇和醋酸钠中,比较分析发现了3个编码烷烃氧化的基因alkMa、alkMb和almA,其中alkMb的差异表达最显著,参与了十二烷的氧化。与醋酸钠培养基上生长的菌株比较,筛选出1 074个差异表达基因,其中622个基因上调,这些基因与烷烃的分解代谢和应激反应有关[28]。红黏土促进A. oleivorans DR1降解十六烷,其参与氧化应激防御相关及上下游代谢的基因上调,表明红黏土提高DR1十六烷利用过程中产生的氧化应激的反应[29]。A. sp. HZ01可以降解蒽、菲、芘、正构烷烃等,利用多环芳烃作为唯一碳源。菌株HZ01在石油处理16 h,转录组分析筛选到742个差异基因,大部分下调基因与细胞运动性、糖代谢和编码核糖体蛋白有关。而脂肪酸代谢途径和部分单加氧酶、脱氢酶被激活,但TCA循环不活跃。末端氧化是菌株HZ01降解正构烷烃的主要好氧途径[30]。
2.2 致病菌的转录组研究开展原核生物转录组研究最多的是致病菌(表 3),转录组已经成为研究宿主-病原菌相互作用复杂性的有效方法。深入了解致病菌的代谢途径从而更好地实时控制,致病菌耐药机制的研究对新药研发提供有力依据。转录组分析发现链霉素依赖菌株Mycobacterium tuberculosis 18b可以作为LTBI模型,并为结核杆菌的进化和核糖体的功能提供了见解[52]。转录组分析了鼠伤寒沙门氏菌Salmonella Typhimurium对氟喹诺酮的耐药机制,耐药性菌株TN4和TW4他们出现了大量差异表达基因,其中28个与耐药相关,26个与两个耐药菌株的双组分系统相关,TW4的输出泵基因差异表达比TN4更强烈[53]。在体外培养条件下鳗弧菌Vibrio anguillarum M3生长中后期代谢途经相关的基因有3 159个,其中129个为新基因[54]。对鳗弧菌转录组研究增强了我们对于其致病力的了解。耐寒产单核李斯特菌Listeria monocytogenes转录组和代谢组分析发现参与合成支链脂肪酸的9个基因及内蛋白A、D基因等都表达上调[55]。转录数据提供了关于L.monocytogenes单核细胞基因冷诱导膜成分变化、其冷应激调控因子生长阶段依赖性以及反义转录本在调节其冷应激反应中的积极作用的新信息。该研究增加了我们对李斯特菌的冷应激反应知识的了解,有助于在食品供应链中对其进行更有效的控制。

转录组是研究表达的RNA转录本功能的,对于解密基因组的功能复杂性及更好地理解生物体的活动,包括发育、免疫防御和应激反应是至关重要的。目前,对于细菌的生理机制研究已扩展到分子层面,利用转录组分析的结果,挖掘出降解功能基因及其代谢通路,构建基因工程菌,进一步提高生物修复的效率。随着基因组学及转录组学研究日益积累,研究人员利用组学数据实现了从更深层面对细菌作用机制的探索。另外,数据共享也加速推进组学研究。近年来对于极端环境的探索也在不断开拓,极端环境存在着丰富的微生物资源,有望在未来的研究中发现更多的微生物,并通过转录组层面研究,对其生长代谢机理进行更深层面的挖掘。目前,高通量测序技术已经发展到第三代,相信在不远的将来,转录组研究会越来越普遍,为科学研究带来帮助。
| [1] |
Piétu G, Eveno E, Sourysegurens B, et al. The genexpress image knowledge base of the human muscle transcriptome: a resource of structural, functional, and positional candidate genes for muscle physiology and pathologies[J]. Genome Research, 1999, 9(12): 1313-1320. DOI:10.1101/gr.9.12.1313 |
| [2] |
Filiatrault MJ. Progress in prokaryotic transcriptomics[J]. Current Opinion in Microbiology, 2011, 14(5): 579-586. DOI:10.1016/j.mib.2011.07.023 |
| [3] |
Roane TM, Pepper IL, Gentry TJ. Microorganisms and metal pollutants[J]. Environmental Microbiology, 2015, 415-439. |
| [4] |
Wenjing L, Yanan W, Chuanyong J. Transcriptome analysis of silver, palladium, and selenium stresses in Pantoea sp. IMH[J]. Chemosphere, 2018, 208: 50-58. DOI:10.1016/j.chemosphere.2018.05.169 |
| [5] |
Zhang X, Wu W, Virgo N, et al. Global transcriptome analysis of hexavalent chromium stress responses in Staphylococcus aureus LZ-01[J]. Ecotoxicology, 2014, 23(8): 1534-1545. DOI:10.1007/s10646-014-1294-7 |
| [6] |
Chourey K, Thompson MR, Morrell-Falvey J, et al. Global molecular and morphological effects of 24-hour chromium (Ⅵ) exposure on Shewanella oneidensis MR-1[J]. Applied and Environmental Microbiology, 2006, 72(9): 6331-6344. DOI:10.1128/AEM.00813-06 |
| [7] |
Dávila Costa JS, Kothe E, Abate CM, et al. Unraveling the Amycolatopsis tucumanensis copper-resistome[J]. Biometals, 2012, 25(5): 905-917. DOI:10.1007/s10534-012-9557-3 |
| [8] |
Clauss-Lendzian E, Vaishampayan A, De JA, et al. Stress response of a clinical Enterococcus faecalis isolate subjected to a novel antimicrobial surface coating[J]. Microbiological Research, 2018, 207: 53-64. DOI:10.1016/j.micres.2017.11.006 |
| [9] |
Saulou-Bérion C, Ignacio G, Brice E, et al. Escherichia coli under ionic silver stress: an integrative approach to explore transcriptional, physiological and biochemical responses[J]. PLoS One, 2015, 10(12): e0145748. DOI:10.1371/journal.pone.0145748 |
| [10] |
Begoña Águila-Clares, Castiblanco LF, José Manuel Quesada, et al. Transcriptional response of Erwinia amylovora upon copper shock: in vivo role of the copA gene[J]. Molecular Plant Pathology, 2017, 19(1): 169-179. |
| [11] |
Gault M, Effantin G, Rodrigue A. Ni exposure impacts the pool of free Fe and modifies DNA supercoiling via metal-induced oxidative stress in Escherichia coli K-12[J]. Free Radical Biology and Medicine, 2016, 97: 351-361. DOI:10.1016/j.freeradbiomed.2016.06.030 |
| [12] |
Vannini A, Pinatel E, Costantini PE, et al. Comprehensive mapping of the Helicobacter pylori NikR regulon provides new insights in bacterial nickel responses[J]. Scientific Reports, 2017, 7: 45458. DOI:10.1038/srep45458 |
| [13] |
Gu W, Semrau JD. Copper and cerium-regulated gene expression in Methylosinus trichosporium OB3b[J]. Applied Microbiology & Biotechnology, 2017, 101(23-24): 8499-8516. |
| [14] |
Marcus SA, Sidiropoulos SW, Steinberg H, et al. CsoR is essential for maintaining copper homeostasis in Mycobacterium tuberculosis[J]. PLoS One, 2016, 11(3): e0151816. DOI:10.1371/journal.pone.0151816 |
| [15] |
Larsen Ø, Karlsen OA. Transcriptomic profiling of Methylococcus capsulatus(Bath)during growth with two different methane monooxygenases[J]. Microbiologyopen, 2016, 5(2): 254-267. DOI:10.1002/mbo3.2016.5.issue-2 |
| [16] |
Quintana J, Novoaaponte L, Argüello JM. Copper homeostasis networks in the bacterium Pseudomonas aeruginosa[J]. Journal of Biological Chemistry, 2017, 292(38): 15691-15704. DOI:10.1074/jbc.M117.804492 |
| [17] |
Zhang H, Ma Y, Liu P, et al. Multidrug resistance operon emrAB contributes for chromate and ampicillin co-resistance in a Staphylococcus strain isolated from refinery polluted river bank[J]. Springerplus, 2016, 5(1): 1648-1659. DOI:10.1186/s40064-016-3253-7 |
| [18] |
Irfan M, Sulman S, Kuipers OP, et al. Ni2+ dependent and psaR mediated regulation of the virulence genes pcpA, psaBCA, and prtA in Streptococcus pneumoniae[J]. PLoS One, 2015, 10(11): e0142839. DOI:10.1371/journal.pone.0142839 |
| [19] |
Chen Y, Huang C, Bai C, et al. Benzo[α]pyrene repressed DNA mismatch repair in human breast cancer cells[J]. Toxicology, 2013, 304: 167-172. DOI:10.1016/j.tox.2013.01.003 |
| [20] |
Brändli RC, Bucheli TD, Ammann S, et al. Critical evaluation of PAH source apportionment tools using data from the Swiss soil monitoring network[J]. Journal of Environmental Monitoring, 2008, 10(11): 1278-1286. DOI:10.1039/b807319h |
| [21] |
Nguyen VD, Wolf C, Mäder U, et al. Transcriptome and proteome analyses in response to 2-methylhydroquinone and 6-brom-2-vinyl-chroman-4-on reveal different degradation systems involved in the catabolism of aromatic compounds in Bacillus subtilis[J]. Proteomics, 2007, 7(9): 1391-1408. DOI:10.1002/(ISSN)1615-9861 |
| [22] |
Atago Y, Shimodaira J, Araki N, et al. Identification of novel extracellular protein for PCB/biphenyl metabolism in Rhodococcus jostii RHA1[J]. Journal of the Agricultural Chemical Society of Japan, 2016, 80(5): 1012-1019. |
| [23] |
Brune I, Becker A, Paarmann D, et al. Under the influence of the active deodorant ingredient 4-hydroxy-3-methoxybenzyl alcohol, the skin bacterium Corynebacterium jeikeium moderately responds with differential gene expression[J]. Journal of Biotechnology, 2006, 127(1): 21-33. |
| [24] |
Cheng Y, Zang H, Wang H, et al. Global transcriptomic analysis of, Rhodococcus erythropolis D310-1 in responding to chlorimuron-ethyl[J]. Ecotoxicology and Environmental Safety, 2018, 157: 111-120. DOI:10.1016/j.ecoenv.2018.03.074 |
| [25] |
Das N, Chandran P. Microbial degradation of petroleum hydrocarbon contaminants: An overview[J]. Biotechnology Research International, 2010, 2011(1): 1-13. |
| [26] |
Wu XL, Yu SL, Gu J, et al. Filomicrobium insigne sp. nov. isolated from an oil-polluted saline soil[J]. International Journal of Systematic & Evolutionary Microbiology, 2009, 59(2): 300-305. |
| [27] |
Van Beilen JB, Funhoff EG. Alkane hydroxylases involved in microbial alkane degradation[J]. Applied Microbiology & Biotechnology, 2007, 74(1): 13-21. |
| [28] |
Kothari A, Charrier M, Wu YW, et al. Transcriptomic analysis of the highly efficient oil-degrading bacterium Acinetobacter venetianus RAG-1 reveals genes important in dodecane uptake and utilization[J]. FEMS Microbiology Letters, 2016, 363(20): fnw224. DOI:10.1093/femsle/fnw224 |
| [29] |
Jung J, Jang IA, Ahn S, et al. Molecular mechanisms of enhanced bacterial growth on hexadecane with red clay[J]. Microbial Ecology, 2015, 70(4): 912-921. |
| [30] |
Hong YH, Deng MC, Xu XM, et al. Characterization of the transcriptome of Achromobacter sp. HZ01 with the outstanding hydrocarbon-degrading ability[J]. Gene, 2016, 584(2): 185-194. |
| [31] |
Scoma A, Barbato M, Hernandez-Sanabria E, et al. Microbial oil-degradation under mild hydrostatic pressure(10 MPa): which pathways are impacted in piezosensitive hydrocarbonoclastic bacteria?[J]. Scientific Reports, 2016, 6: 23526. DOI:10.1038/srep23526 |
| [32] |
Jin HM, Jeong HI, Kim KH, et al. Genome-wide transcriptional responses of Alteromonas naphthalenivorans SN2 to contaminated seawater and marine tidal flat sediment[J]. Scientific Reports, 2016, 6(1): 21796. DOI:10.1038/srep21796 |
| [33] |
Cruz A, Rodrigues R, Pinheiro M, et al. Transcriptomes analysis of Aeromonas molluscorum Av27 cells exposed to tributyltin(TBT): Unravelling the effects from the molecular level to the organism[J]. Marine Environmental Research, 2015, 109: 132-139. DOI:10.1016/j.marenvres.2015.06.017 |
| [34] |
Scheublin TR, Deusch S, Morenoforero SK, et al. Transcriptional profiling of Gram-positive Arthrobacter in the phyllosphere: induction of pollutant degradation genes by natural plant phenolic compounds[J]. Environmental Microbiology, 2014, 16(7): 2212-2225. DOI:10.1111/emi.2014.16.issue-7 |
| [35] |
Kun W, Hua Y, Hui P, et al. Bioremediation of triphenyl phosphate in river water microcosms: Proteome alteration of Brevibacillus brevis and cytotoxicity assessments[J]. Science of The Total Environment, 2019, 649: 563-570. DOI:10.1016/j.scitotenv.2018.08.342 |
| [36] |
Patrauchan MA, Parnell JJ, Mcleod MP, et al. Genomic analysis of the phenylacetyl-CoA pathway in Burkholderia xenovorans LB400[J]. Archives of Microbiology, 2011, 193(9): 641-650. DOI:10.1007/s00203-011-0705-x |
| [37] |
Peng X, Yamamoto S, Alain A. Vertès, et al. Global transcriptome analysis of the tetrachloroethene-dechlorinating bacterium Desulfito bacterium hafniense Y51 in the presence of various electron donors and terminal electron acceptors[J]. Journal of Industrial Microbiology & Biotechnology, 2012, 39(2): 255-268. |
| [38] |
Johnson DR, Nemir A, Andersen GL, et al. Transcriptomic microarray analysis of corrinoid responsive genes in Dehalococcoides ethenogenes strain 195[J]. FEMS Microbiology Letters, 2010, 294(2): 198-206. |
| [39] |
Johnson DR, Brodie EL, Hubbard AE, et al. Temporal transcriptomic microarray analysis of Dehalococcoides ethenogenes strain 195 during the transition into stationary phase[J]. Applied & Environmental Microbiology, 2008, 74(9): 2864-2872. |
| [40] |
Lee J, Hiibel SR, Reardon KF, et al. Identification of stress-related proteins in Escherichia coli using the pollutant cis-dichloroethylene[J]. J Appl Microbiol, 2010, 108(6): 2088-2102. |
| [41] |
Hristova KR, Schmidt R, Chakicherla AY, et al. Comparative transcriptome analysis of Methylibium petroleiphilum PM1 exposed to the fuel oxygenates methyl tert-butyl ether and ethanol[J]. Applied & Environmental Microbiology, 2007, 73(22): 7347-7357. |
| [42] |
Fida TT, Moreno-Forero SK, Breugelmans P, et al. Physiological and transcriptome response of the polycyclic aromatic hydrocarbon degrading Novosphingobium sp. LH128 after inoculation in soil[J]. Environmental Science & Technology, 2017, 51(3): 1570-1579. |
| [43] |
Guan X, Liu F, Wang J, et al. Mechanism of 1, 4-dioxane microbial degradation revealed by 16S rRNA and metatranscriptomic analyses[J]. Water Science & Technology A Journal of the International Association on Water Pollution Research, 2018, 77(1): 123-133. |
| [44] |
Chen Q, Tu H, Luo X, et al. The regulation of para-nitrophenol degradation in Pseudomonas putida DLL-E4[J]. PLoS One, 2016, 11(5): e0155485. DOI:10.1371/journal.pone.0155485 |
| [45] |
Dubey SK, Tokashiki T, Suzuki S. Microarray-mediated transcriptome analysis of the tributyltin(TBT)-resistant bacterium Pseudomonas aeruginosa 25W in the presence of TBT[J]. Journal of Microbiology, 2006, 44(2): 200-205. |
| [46] |
Muller JF, Stevens AM, Craig J, et al. Transcriptome analysis reveals that multidrug efflux genes are upregulated to protect Pseudomonas aeruginosa from pentachlorophenol stress[J]. Applied & Environmental Microbiology, 2007, 73(14): 4550-4558. |
| [47] |
Yang X, Xi J. Transcriptomic and benzoate metabolic pathways of Rhodococcus sp. R04 cultured in biphenyl[J]. Acta Microbiologica Sinica, 2015, 55(7): 851-862. |
| [48] |
Edmilson R, Gonçalves, Hara H, Miyazawa D, et al. Transcriptomic assessment of isozymes in the biphenyl pathway of Rhodococcus sp. strain RHA1[J]. Applied & Environmental Microbiology, 2006, 72(9): 6183-6193. |
| [49] |
Sharp JO, Sales CM, Leblanc JC, et al. An inducible propane monooxygenase is responsible for n-nitrosodimethylamine degradation by Rhodococcus sp. strain RHA1[J]. Applied and Environmental Microbiology, 2007, 73(21): 6930-6938. DOI:10.1128/AEM.01697-07 |
| [50] |
Benli C, Tsoi TV, Shoko I, et al. Sphingomonas wittichii strain RW1 genome-wide gene expression shifts in response to dioxins and clay[J]. PLoS One, 2016, 11(6): e0157008. DOI:10.1371/journal.pone.0157008 |
| [51] |
Morenoforero SK, Meer JRVD. Genome-wide analysis of Sphingomonas wittichii RW1 behaviour during inoculation and growth in contaminated sand[J]. Isme Journal, 2014, 9(1): 150-165. |
| [52] |
Benjak A, Uplekar S, Zhang M, et al. Genomic and transcriptomic analysis of the streptomycin-dependent Mycobacterium tuberculosis strain 18b[J]. Bmc Genomics, 2016, 17(1): 190-204. DOI:10.1186/s12864-016-2528-2 |
| [53] |
Lin L, Dai X, Ying W, et al. RNA-seq-based analysis of drug-resistant Salmonella enterica serovar Typhimurium selected in vivo and in vitro[J]. PLoS One, 2017, 12(4): e0175234. DOI:10.1371/journal.pone.0175234 |
| [54] |
李贵阳, 莫照兰, 李杰.海水病原鳗弧菌M3体外转录表达分析[C]中国水产学会鱼病专业委员会2013年学术研讨会论文摘要汇编. 2013.
|
| [55] |
Hingston P, Chen J, Allen K, et al. Strand specific RNA-sequencing and membrane lipid profiling reveals growth phase-dependent cold stress response mechanisms in Listeria monocytogenes[J]. PLoS One, 2017, 12(6): e0180123. DOI:10.1371/journal.pone.0180123 |
| [56] |
Willig CJ, Duan K, Zhang ZJ. Transcriptome profiling of plant genes in response to Agrobacterium tumefaciens-mediated transformation[J]. 2018, 418: 319-348.
|
| [57] |
Carlson PE, Bourgis AET, Hagan AK, et al. Global gene expression by Bacillus anthracis during growth in mammalian blood[J]. Pathogens and Disease, 2015, 73(8): ftv061. DOI:10.1093/femspd/ftv061 |
| [58] |
Manuel W, Tobias B, Gaspar AH, et al. Transcriptome sequencing of the human pathogen Corynebacterium diphtheriae NCTC 13129 provides detailed insights into its transcriptional landscape and into DtxR-mediated transcriptional regulation[J]. BMC Genomics, 2018, 19(1): 82-99. DOI:10.1186/s12864-018-4481-8 |
| [59] |
Kathleen N, Ryan MC, Matthew M, et al. Transcriptome Analysis of Neisseria gonorrhoeae during natural infection reveals differential expression of antibiotic resistance determinants between men and women[J]. Msphere, 2018, 3(3): e00312-e00318. |
| [60] |
Cruz-Rabadán JS, Miranda-Ríos J, Espín-Ocampo G, et al. Non-coding RNAs are differentially expressed by nocardia brasiliensis in vitro and in experimental actinomycetoma[J]. Open Microbiology Journal, 2017, 11: 112-125. DOI:10.2174/1874285801711010112 |
| [61] |
Machuca A, Martinez V. Transcriptome analysis of the intracellular facultative pathogen Piscirickettsia salmonis: expression of putative groups of genes associated with virulence and iron metabolism[J]. PLoS One, 2016, 11(12): e0168855. DOI:10.1371/journal.pone.0168855 |
| [62] |
Marrer E, Satoh AT, Johnson MM, et al. Global transcriptome analysis of the responses of a fluoroquinolone-resistant Streptococcus pneumoniae mutant and its parent to ciprofloxacin[J]. Antimicrobial Agents & Chemotherapy, 2006, 50(1): 269-278. |
| [63] |
马月姣.酸耐受副溶血性弧菌生物学特性及转录组、蛋白组分析[D].上海: 上海海洋大学, 2016. http://cdmd.cnki.com.cn/Article/CDMD-10264-1016912179.htm
|
| [64] |
Sánchez J, Holmgren J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects[J]. Cellular & Molecular Life Sciences, 2008, 65(9): 1347-1360. |
| [65] |
陈保立.霍乱弧菌活的非可培养状态的转录组及蛋白组研究[D].北京: 中国疾病预防控制中心, 2013. http://cdmd.cnki.com.cn/article/cdmd-84501-1015525971.htm
|
| [66] |
Chen X, Sun C, Laborda P, et al. Melatonin treatment inhibits the growth of Xanthomonas oryzae pv. oryzae[J]. Frontiers in Microbiology, 2019, 9: 2280. |