




2. 大同市第二人民医院肿瘤内科,大同 037000
2. Department of Medical Oncology, the Second People's Hospital of Datong, Datong 037000
原发性肝癌的发病率居全球第5位,致死率排第3位,其中以肝细胞癌(Hepatocellular carcinoma,HCC)最为常见[1]。生物分子间彼此相互作用形成的网络体系是生物进程运行的基础。网络的失调是导致多种疾病尤其是肿瘤等重大疾病的根本原因。在这些网络模体中最为普遍与重要的是前馈环路(Feed-forward loops,FFLs)[2]。FFL由两个输入调节因子P1与P2,以及P1与P2共同调节的靶因子P3组成。FFL分连贯FFL(Coherent FFL)与非连贯FFL(Incoherent FFL)两类[3]。连贯FFL指P1调控P2、P3和P2调控P3均为正向(Positive)调控,而非连贯FFL是指P1调控P2、P3为正向,而P2调控P3为负向(Negative)调控。
增强子(Enhancer)一般是几百碱基对长度的DNA片段,并能被多个转录因子占据,在基因调控中通过顺式调控原件对靶基因起正调控作用。已有研究表明肝细胞癌中的增强子突变会导致增强子失活,进而影响靶基因的表达[4]。MicroRNA(miRNA)是一类长度为18-24 nt的非编码小RNA,在进化过程中高度保守,它通过与靶基因的3' UTR区特异性结合从而在转录后水平抑制靶基因的表达或直接降解靶mRNA,异常表达的miRNA在肝癌的发病机制中起重要作用。先前研究发现,增强子调控miRNA参与肿瘤的发生与发展,从而使得增强子、miRNA与转录因子可形成重要的调控单位FFL[5]。那么增强子与miRNA是否可以形成FFL目前还并不清楚。本课题组通过生物信息学手段进行了增强子调控miRNA的识别,并筛选了其参与的FFL,对已通过实验验证的miRNA靶基因分析,基因注释后发现上述的FFL参与多种与肝癌相关的信号通路或生物进程。本研究旨在通过对FFL的识别与分析为以FFL为网络模体单元的肝肿瘤调控机制以及肝肿瘤标志物识别方面奠定前期工作基础。
1 材料与方法 1.1 HepG2细胞系中增强子的识别从ENCODE[6]数据库下载得到HepG2细胞系的DNase高敏位点以及H3K4me1、H3K27ac、H3K4me3三类组蛋白修饰信息的ChIP-seq数据,根据增强子特征性的组蛋白修饰信号预测HepG2细胞系中的增强子。识别增强子区域基于如下特征[7-8]:(1)增强子区域中心位置存在DNase高敏位点; (2)在增强子区域内存在明显的H3K4me1和H3K27ac信号,且呈峰-谷-峰的趋势; (3)增强子区域内的H3K4me1信号比H3K4me3信号强。
1.2 HepG2细胞系中转录因子-增强子-miRNA FFL的识别 1.2.1 增强子-miRNA调控关系的识别从GENCODE[9]数据库得到蛋白编码基因的注释信息,提取得到蛋白编码基因的转录起始位点信息。从FANTOM5[10]数据库获取miRNA的转录起始位点信息。根据Suzuki等[5]的识别方法,对每个增强子分别找到其距离最近的miRNA转录起始位点及同方向(同在增强子上游或下游)的蛋白编码基因的转录起始位点。将增强子中心与最近的miRNA转录起始位点的距离记为M,增强子中心与同方向基因转录起始位点的距离记为G,根据公式(1)计算Score值,以0.2为阈值,设定Score得分在0-0.2范围内的增强子与miRNA为可能存在的调控关系。
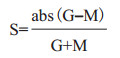 (1)
(1)
从ENCODE数据库中下载得到HepG2细胞系中miRNA的表达量信息,据此对上述调控关系进行筛选,只有涉及在HepG2细胞系中表达的miRNA的调控关系被采用为最后的结果。
1.2.2 转录因子-增强子调控关系的识别从UCSC及CistromeDB[11]数据库中下载得到HepG2细胞系中65个转录因子的ChIP-seq数据,作为转录因子结合位点的信息。如果转录因子结合位点落在增强子区间内,则认为该转录因子与该增强子存在调控关系。
1.2.3 转录因子-miRNA调控关系的识别若1.2.2中得到的转录因子结合位点信息落在miRNA转录起始位点上游10 kb~下游1 kb范围内,则认为该转录因子对于该miRNA存在调控关系[12]。
1.2.4 转录因子-增强子-miRNA FFL的识别根据1.2.1 1.2.2 1.2.3中的调控关系得到转录因子-增强子-miRNA FFL。对于所得结果进行超几何检验,如公式(2)所示,M、N和k分别代表miRNA总数、细胞中所有受到转录因子调控的miRNA数量、细胞中受某一转录因子调控的miRNA数量,所得p值越小,说明对应的转录因子和增强子共同调控的miRNA数量越多。随后,通过Benjamini-Hochberge[13]方法,根据p值得到FDR值,设定q值在0-0.05范围内的为非随机出现的FFL,即所得最终结果。
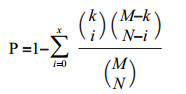 (2)
(2)
根据所得的FFL统计涉及的每个miRNA参与的FFL数目,得到其中参与FFL数目明显高于总体平均值的miRNA(参与FFL数大于上界,在所有miRNA的参与FFL数中属于上离群点范围),定义为FFL中的核心miRNA。
1.3.2 靶基因预测从TarBase[14],mirTarBase[15]数据库下载得到实验验证的miRNA的靶基因信息。分别得到2.3.1中核心miRNA的靶基因。
1.3.3 功能富集分析利用R软件包clusterProfiler[16],对于上述得到的所有靶基因做Gene Ontology(GO)[17]、KEGG Pathway[18]富集分析。
2 结果 2.1 增强子识别根据DNase高敏位点及组蛋白修饰,最终得到5 055个增强子。这些增强子的DNase及3类组蛋白修饰信号分布趋势,如图 1所示。

|
| 图 1 DNase及组蛋白修饰信号 |
从图 1可知增强子中心上下游1 kb存在显著的DNase活性。另外,在增强子区域存在高的H3K27ac信号,以及高的H3K4me1信号与低的H3K4me3信号。
2.2 转录因子-增强子-miRNA FFL通过超几何检验,过滤q值小于0.05的结果后,我们最终得到2 070个FFL,因增强子调控miRNA以正调控为主,因为本文识别的增强子为连贯FFL。这些FFL共涉及57个转录因子,180个增强子,85个miRNA。
2.3 功能富集分析结果利用R软件包clusterProfiler[16],我们对识别的2 070个FFL的miRNA涉及的靶基因进行了GO[17]与KEGG[18]功能富集分析,结果(图 2)表明FFL中miRNA的靶基因显著富集于肝癌相关的信号通路或生物进程。如病毒致癌通路、p53信号通路、细胞周期相关的通路、细胞周期阻滞等。

|
| 图中左右两列分别列出了核心miRNA的靶基因最为显著富集的10条Gene Ontology进程及KEGG通路 图 2 参与FFL的核心miRNA靶基因的基因功能注释结果 |
在2 070个FFL涉及的85个miRNA中,有5个miRNA在肿瘤和正常样本中存在表达差异(|log2(FC)| > 1,p < =0.05),分别为hsa-miR-455(|log2(FC)|:2.803),hsa-miR-224(|log2(FC)|:3.615),hsa-miR-452(|log2(FC)|:3.111),hsa-miR-10b(|log2(FC)|:2.989),hsa-miR-574(|log2(FC)|:2.799)。此外,从绝对表达量来讲,上述5个miRNA中hsa-miR-574和hsa-miR-92a的表达量(CPM)显著高于85个总miRNA的表达量平均值(85个miRNA在肝癌细胞中CPM平均值7.48,hsa-miR-574的CPM为129.96,hsa-miR-92a的CPM为98.81)。
结果显示hsa-miR-574在HepG2中显著参与了多的FFL(平均每个miRNA参与24个FFL,hsa-miR-574参与的FFL数量为99个,图 3),这在hsa-miR-574参与的99个FFL中共涉及4个增强子(chr4:38160530-38164680,chr4:38162070-38166220,chr4:38179330-38184380,chr4:38223510-38227660)和29个转录因子。在这些转录因子中,有16个参与了4个FFL(NFIC,MAX,HNF4G,RAD21,ARID3A,TAF1,CREB1,HDAC2,MYBL2,FOXA2,HNF4A,RXRA,FOXA1,JUND,FOSL2,SP1),为参与FFL的数目最多。而在4个增强子中,chr4:38162070-38166220参与的调控FFL数量最多,为29个。

|
| 图 3 涉及hsa-miR-574的FFL构成的网络图 |
为此,我们对这一个miRNA的靶基因进行了KEGG通路富集分析,结果(图 4)显示hsa-miR-574的靶基因显著与多个肿瘤相关的信号通路有关。其中,富集最为显著的通路——cAMP信号通路被已有文献证明可以抑制肝癌细胞增殖而促进其分化[19]。

|
| 图 4 hsa-miR-574靶基因的KEGG通路富集结果 |
根据我们识别的5 055个增强子的组蛋白修饰信号分布来看,DNase信号显著富集在增强子中心附近,这与活性增强子的染色体开放特征一致。另外,这些增强子体现出高的H3K27ac信号以及高的H3K4me1/H3K4me3占比,这与先前文献报道的活性增强子信号特征一致。最终我们识别了2 070个FFL,其中65个转录因子有57个参与了FFL(占88%),而相比转录因子而言,增强子和miRNA参与的FFL比例要明显低于转录因子,这一结果说明在FFL中,转录因子起到广泛结合的作用,而增强子与miRNA由于其特异性导致参与的FFL相对较少。在功能富集方面,结果表明FFL中miRNA的靶基因显著富集于肝癌相关的信号通路或生物进程中,这些结果说明我们基于肝癌组学数据识别的FFL显著与肿瘤相关,这也验证了我们识别的FFL的有效性。此外,除了从表达正常与肿瘤组织的表达差异与绝对表达量多少来衡量miRNA参与肿瘤的重要性外,对于处于FFL网络中的miRNA来说,miRNA参与FFL的频率是考量miRNA在肿瘤网络中是否起关键作用的另一重要因素。因此,我们重点考察了hsa-miR-574在网络中的出现频率,结果显示hsa-miR-574参与的99个FFL,较平均的24个FFL显著高,并且功能富集分析也发现hsa-miR-574的靶基因显著与多个肿瘤相关的信号通路有关。之前已有文献表明,肝癌患者体细胞中hsa-miR-574的表达量显著高于正常样本,由此推测该miRNA可以作为肝癌诊断的肿瘤标记物,这与我们的结果相符合[20]。研究结果初步探讨了以增强子-miRNA为核心的FFL在肝细胞癌中的特征与功能,有望为基于网络模体为单位的肝肿瘤标志物识别奠定理论与数据基础。
4 结论本文基于肝癌细胞系HepG2中的DNase高敏位点以及H3K4me1、H3K27ac、H3K4me3组蛋白修饰特征这些普遍认可的表观遗传学特征为识别活性增强子的理论基础,识别共得到5 055个肝癌特异的增强子。通过处理增强子与miRNA位置信息,以及65个转录因子的ChIP-seq数据获得转录因子结合位点,构建了增强子-miRNA、转录因子-miRNA与转录因子-miRNA调控关系。通过超几何检验筛选了2 070个FFL。其中共涉及57个转录因子,180个增强子与85个miRNA。GO与KEGG功能富集分析FFL的miRNA靶基因显示这些靶基因广泛的参与了与肝癌的发生发展相关的生物学进程与调控通路。
| [1] |
Yang JD, Roberts LR. Epidemiology and management of hepatocellular carcinoma[J]. Infect Dis Clin N Am, 2010, 24(4): 899-919. DOI:10.1016/j.idc.2010.07.004 |
| [2] |
Alon U. Network motifs:theory and experimental approaches[J]. Nature Reviews Genetics, 2007, 8(6): 450-61. DOI:10.1038/nrg2102 |
| [3] |
Mangan S, Alon U. Structure and function of the feed-forward loop network motif[J]. Proc Natl Acad Sci USA, 2003, 100(21): 11980-11985. DOI:10.1073/pnas.2133841100 |
| [4] |
Huang D, Ovcharenko I. Identifying causal regulatory SNPs in ChIP-seq enhancers[J]. Nucleic Acids Res, 2015, 43(1): 225-236. DOI:10.1093/nar/gku1318 |
| [5] |
Suzuki HI, Young RA, Sharp PA. Super-enhancer-mediated RNA processing revealed by integrative microRNA network analysis[J]. Cell, 2017, 168(6): 1000-1014. DOI:10.1016/j.cell.2017.02.015 |
| [6] |
Feingold EA, Good PJ, Guyer MS, et al. The ENCODE(ENCyclopedia of DNA elements)project[J]. Science, 2004, 306(5696): 636-640. DOI:10.1126/science.1105136 |
| [7] |
Elkon R, Agami R. Characterization of noncoding regulatory DNA in the human genome[J]. Nat Biotechnol, 2017, 35(8): 732-746. DOI:10.1038/nbt.3863 |
| [8] |
Long HK, Prescott SL, Wysocka J. Ever-changing landscapes:transcriptional enhancers in development and evolution[J]. Cell, 2016, 167(5): 1170-1187. DOI:10.1016/j.cell.2016.09.018 |
| [9] |
Harrow J, Frankish A, Gonzalez JM, et al. GENCODE:The reference human genome annotation for The ENCODE Project[J]. Genome Res, 2012, 22(9): 1760-1774. DOI:10.1101/gr.135350.111 |
| [10] |
De Rie D, Abugessaisa I, Alam T, et al. An integrated expression atlas of miRNAs and their promoters in human and mouse[J]. Nat Biotechnol, 2017, 35(9): 872-878. DOI:10.1038/nbt.3947 |
| [11] |
Mei SL, Qin Q, Wu Q, et al. Cistrome Data Browser:a data portal for ChIP-Seq and chromatin accessibility data in human and mouse[J]. Nucleic Acids Res, 2017, 45(D1): D658-D662. DOI:10.1093/nar/gkw983 |
| [12] |
Chang LW, Viader A, Varghese N, et al. An integrated approach to characterize transcription factor and microRNA regulatory networks involved in Schwann cell response to peripheral nerve injury[J]. BMC Genomics, 2013, 14: 84. DOI:10.1186/1471-2164-14-84 |
| [13] |
Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing[J]. J R Stat Soc B, 1995, 57(1): 289-300. |
| [14] |
Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, et al. DIANA-TarBase v8:a decade-long collection of experimentally supported miRNA-gene interactions[J]. Nucleic Acids Res, 2018, 46(D1): D239-D245. DOI:10.1093/nar/gkx1141 |
| [15] |
Chou CH, Shrestha S, Yang CD, et al. miRTarBase update 2018:a resource for experimentally validated microRNA-target interac-tions[J]. Nucleic Acids Res, 2018, 46(D1): D296-D302. DOI:10.1093/nar/gkx1067 |
| [16] |
Yu GC, Wang LG, Han YY, et al. clusterProfiler:an R package for comparing biological themes among gene clusters[J]. Omics, 2012, 16(5): 284-287. DOI:10.1089/omi.2011.0118 |
| [17] |
The Gene Ontology C. Expansion of the gene ontology knowledge-base and resources[J]. Nucleic Acids Res, 2017, 45(D1): D331-D338. DOI:10.1093/nar/gkw1108 |
| [18] |
Kanehisa M, Furumichi M, Tanabe M, et al. KEGG:new perspectives on genomes, pathways, diseases and drugs[J]. Nucleic Acids Res, 2017, 45(D1): D353-D361. DOI:10.1093/nar/gkw1092 |
| [19] |
Ionta M, Rosa MC, Almeida RB, et al. Retinoic acid and cAMP inhibit rat hepatocellular carcinoma cell proliferation and enhance cell differentiation[J]. Braz J Med Biol Res, 2012, 45(8): 721-729. DOI:10.1590/S0100-879X2012007500087 |
| [20] |
Shen X, Xue Y, Cong H, et al. Dysregulation of serum microRNA-574-3p and its clinical significance in hepatocellular carcinoma[J]. Ann Clin Biochem, 2018, 55(4): 478-484. DOI:10.1177/0004563217741908 |