




2. 内蒙古大学省部共建草原家畜生殖调控与繁育国家重点实验室,呼和浩特 010070;
3. 中国农业大学食品科学与营养工程学院,北京 100083
2. State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, Inner Mongolia University, Hohhot 010070;
3. College of Food Science and Nutritional Engineering, China Agricultural University, Beijing 100083
基因编辑技术是通过对目的基因进行编辑,从而实现对基因组精确修饰的一种崭新方法。目前常用的基因编辑方法主要有:锌指核酸酶(Zinc-finger nucleases,ZFNs)[1]、转录激活样效应因子核酸酶(Transcription activator-like effector nucleases,TALEN)[2]和成簇规律间隔短回文重复(Clustered regularly interspaced short palindromic repeats,CRISPR)/ CRISPR相关蛋白9(CRISPR associated protein 9,Cas9)[3],近年来由于CRISPR/Cas9具有高效性、易操作、低成本、周期短等优点[4],在动植物的基因修饰中深受青睐。
在动物中,通过对不利基因编辑使其功能丧失来进行动物的改良。Myostation(MSTN)基因编码的肌肉生长抑制素是一种肌肉生长负调节因子[5-6],动物体内缺失或失活可能导致肌肉细胞数量增加,肌纤维直径增大,肌纤维数量增加,肌肉过度发育,但对动物的生存无影响[7]。自然突变或基因编辑都可能导致MSTN发生功能性失活,而表现“双肌”现象。Hanset等发现自然突变引起“双肌”的比利时蓝牛比普通牛的肌纤维数量更多[8]。近年来内蒙古大学以牛为研究对象成功进行MSTN的编辑使其缺失了6 bp,其中4 bp在第一外显子上,因而形成移码突变,不能产生正常的蛋白质而丧失功能,使编辑牛的产肉量和生长速度得到显著提高。
目前用于基因编辑检测方法主要有:PCR-限制性核酸内切酶(PCR-restriction enzyme,PCR-RE),T7核酸内切酶Ⅰ(T7 endonuclease I,T7EI),Surveyor核酸酶和高分辨率熔解曲线(High-resolution melting,HRM)分析等。PCR-RE法是根据限制性核酸内切酶酶切位点的变化,对突变体进行鉴定以及基因分型的方法[9];T7EI和Surveyor核酸酶是一种非配对内切酶,通过识别并切割异源双链DNA,产生两个或更多个较小的片段,从而达到对突变体进行鉴定的目的[10];HRM分析法是利用与荧光染料结合的双链DNA在温度升高过程中会发生减色效应的物理性质,通过检测双链DNA在溶解过程中释放的染料荧光信号所形成的特征溶解曲线进行基因分型[11]。虽然这些方法可以成功地用于突变体的筛选或基因分型,但是也存在一定的局限性,诸如需要限制性核酸内切酶酶切位点,酶切不完全,成本较高等。由于基因编辑后缺失的核苷酸数量最少是1 bp,也有的是几个bp,所以对于这种小片段的缺失常规PCR方法无法进行检测和分型。功能核酸是指可与特定目标物高选择性结合[12]或具有催化功能[13]的核酸分子,它具有检测、识别、催化、电子传递、发光等用途。因此可以在常规PCR方法基础上引入功能核酸对小片段的缺失进行检测和分型。本研究以MSTN编辑牛为研究材料,建立功能核酸PCR(Functional nucleic acid PCR,FNA-PCR)检测方法,拟对MSTN编辑牛进行检测和基因分型,为基因编辑动物检测提供一种简便的检测方法。
1 材料与方法 1.1 材料本研究所用MSTN编辑牛样品(编辑位点:AC_000159.1:g.1102 del GAGTGT)和对照样品由内蒙古大学提供。
1.2 方法 1.2.1 基因组DNA的提取采用氯仿苯酚法提取样品DNA[14]。DNA浓度由核酸测定仪检测得到。
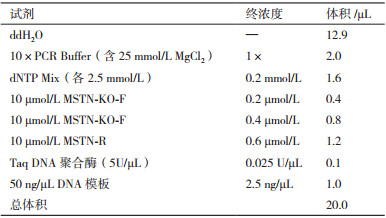
1.2.2 引物设计使用Primer Premier 5.0软件,针对编辑位点的两种等位基因设计两条不同的正向引物和一条共用的反向引物,其中一条正向引物是用于检测野生型等位基因:MSTN-WT-F:5'-ATGCTCGAGGCTTATCTATAGCATTGAAGATTACCATGCCCACGGAGTGT-3',3'端终止于编辑位点,同时对它的5'端进行功能核酸接头设计,即添加29 bp功能核酸序列(ATGCTCGAGGCTTATCTATAGCATTGAAG)。本研究所用的功能核酸是与牛基因组序列不同源的一段核酸序列。另一条正向引物用于检测编辑型等位基因:MSTN-KO-F:5'-ATTACCATGCCCACGGA-GTAG-3',3'端跨越编辑位点向后延伸6 bp。共用的反向引物:MSTN-R:5'-CTCTTTCCCCTCCTCCT-TACA-3',它位于编辑位点下游(图 1)。引物MSTN-WT-F和MSTN-R用于检测野生型等位基因扩增片段为175 bp,MSTN-KO-F和MSTN-R用于检测编辑型等位基因扩增片段为140 bp。引物由北京擎科新业生物技术有限公司合成。

|
| 图 1 引物设计示意图 MSTN-WT-F引物5'端灰色斜线代表功能核酸接头;MSTN-KO-F引物中蓝色短线代表此处碱基不存在;模板中蓝色短线代表此处碱基被编辑 |
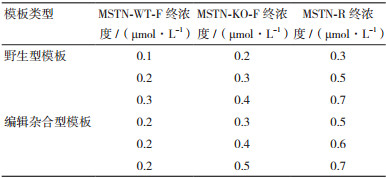
分别以野生型和编辑杂合型(+/-)样品基因组DNA为模板进行PCR反应体系和条件的优化,引物终浓度梯度如表 1,并设置退火温度为60℃、61℃、62℃、63℃ 4个梯度,三条引物添加在同一体系进行扩增。PCR反应程序为:94℃ 5 min;94℃ 30 s,60℃ 30 s,72℃ 30 s,共35个循环;72℃ 5 min。PCR产物用2.5%的琼脂糖凝胶电泳进行鉴定。
将MSTN编辑牛基因组DNA进行稀释得到含25 ng、15 ng、10 ng、1 ng、0.1 ng和0.01 ng的样品,然后按照已经优化好的体系(表 2)进行方法灵敏度测试。
选取5个不同基因型个体,用建立的方法进行鉴定及基因分型。
2 结果 2.1 FNA-PCR反应体系优化将针对编辑型和野生型等位基因设计的正向引物及它们共用的反向引物在同一体系中进行引物浓度和退火温度的优化。当以野生型牛基因组DNA为模板时,在所有条件下都只扩增出一条175 bp目的条带,说明只有引物MSTN-WT-F和MSTN-R发挥作用,引物之间无互作且特异性好(图 2)。且当MSTN-WT-F引物终浓度为0.2 μmol/L、退火温度为60℃时扩增条带最亮;当以编辑杂合(+/-)型牛基因组DNA为模板时,所有条件下都可见两条明显的目的条带(一条是175 bp,另一条是140 bp),且在引物终浓度为MSTN-WT-F:0.2 μmol/L,MSTN-KO-F:0.4 μmol/L,MSTN-R:0.6 μmol/L,退火温度为60℃时,扩增出较亮,更易区分,且引物二聚体相对较弱的目的条带(图 3)。综合两种不同类型模板体系优化结果可知最优条件为:0.2 μmol/L MSTN-WT-F,0.4 μmol/L MSTN-KO-F,0.6 μmol/L MSTN-R的引物终浓度,和60℃退火温度。

|
| 图 2 野生型牛基因组DNA不同引物浓度及退火温度的PCR结果 M:Marker I;1:空白对照;2-5:60℃,61℃,62℃,63℃。A:MSTNWT-F:0.1 μmol/L,MSTN-KO-F:0.2 μmol/L,MSTN-R:0.3 μmol/L。B:MSTN-WT-F:0.2 μmol/L,MSTN-KO-F:0.3 μmol/L,MSTN-R:0.5 μmol/L。C:MSTN-WT-F:0.3 μmol/L,MSTN-KO-F:0.4 μmol/L,MSTN-R:0.7 μmol/L |

|
| 图 3 编辑杂合型(+/-)牛基因组DNA不同引物浓度及退火温度的PCR结果 M:Marker Ⅰ;1:空白对照;2-5:60℃,61℃,62℃,63℃。A:MSTNWT-F:0.2 μmol/L,MSTN-KO-F:0.3 μmol/L,MSTN-R:0.5 μmol/L。B:MSTN-WT-F:0.2 μmol/L,MSTN-KO-F:0.4 μmol/L,MSTN-R:0.6 μmol/L。C:MSTN-WT-F:0.2 μmol/L,MSTN-KO-F:0.5 μmol/L,MSTN-R:0.7 μmol/L |
按照上述已经优化好的体系对含25 ng、15 ng、10 ng、1 ng、0.1 ng和0.01 ng MSTN编辑牛基因组DNA成分样品进行灵敏度检测。结果显示,0.1 ng及以上所有含量梯度均能特异性扩增出预期两条目的片段,即在反应体系中含0.1 ng的MSTN编辑牛基因组DNA就可被检测出,并可对其进行有效鉴别,表明本文中方法的检出限为0.1 ng(图 4)。

|
| 图 4 引物灵敏度检测结果 M:Marker Ⅰ;1:空白对照;2-7分别为:MSTN编辑牛基因组DNA含量分别为25 ng、15 ng、10 ng、1 ng、0.1 ng和0.01 ng |
利用FNA-PCR方法根据优化好的退火温度和引物浓度对5个个体进行鉴定及基因分型,每个个体设置2个重复。检测结果(图 5)显示,所有检测结果中均没有非特异性扩增,条带清晰,且杂合型个体中两条扩增条带大小差异明显。以野生型牛个体DNA为模板只能扩增出一条目的片段(175 bp);以MSTN编辑杂合型(+/-)牛个体DNA为模板可同时扩增出大小为175 bp和140 bp的目的片段,从而实现基因编辑牛和野生型牛个体的有效检测。

|
| 图 5 MSTN编辑牛样品FNA-PCR检测结果 M:Marker Ⅰ;1:空白对照组;2-3:野生型(WT)个体;4-6:编辑杂合型(+/-)牛个体 |
自2008年我国启动转基因生物新品种培育重大专项后,转基因动物如转人乳清白蛋白[15]、人溶菌酶[16]、人乳铁蛋白[17]的转基因牛羊等不断被研发出来并将成为新型的育种材料。由于转入外源基因的转基因动植物一直受到社会质疑,转基因生物的研制方向逐渐向内源基因的编辑方向发展,为此基因编辑技术将成为未来研究的热点。2018年7月25日,欧盟通过了“由基因编辑技术获得的生物品种,将被作为转基因生物(GMO),纳入欧盟严格的转基因监管框架中”的裁决,因此迫切需要建立针对基因编辑产品的检测技术平台,对基因编辑产品进行快速简便的检测。本研究建立的FNA-PCR方法通过一次PCR扩增和琼脂糖凝胶电泳,不仅可以对基因编辑个体进行检测,还可以对其基因型进行判定。FNA-PCR与其它基因编辑生物鉴定或基因分型方法相比具有独特的优势,与PCR-RE方法相比,FNA-PCR不受限制性核酸内切酶酶切位点的限制,而PCR-RE法需要存在酶切位点才可以进行检测[18];与T7EI和Surveyor核酸酶法相比,FNA-PCR能够准确地对基因型进行区分,而T7EI和Surveyor核酸酶法无法实现此目的[19];与本文方法相比,HRM法需要特殊的设备,成本较高[20]。
FNA-PCR反应中引物的设计是获得成功的关键之一。FNA-PCR的引物设计比传统PCR引物的设计更加巧妙,既可对编辑样品进行检测又能对其进行基因分型。因此设计引物时应该考虑以下几个问题:(1)用于检测编辑型等位基因的引物不能在野生型等位基因检测中发挥作用,因此,本研究在正向引物的3'端进行了创新性设计,即两条正向引物中的一条3'端终止于编辑位点,另一条跨越编辑位点再延伸几个bp;(2)由于基因编辑后缺失的片段较小,利用传统的PCR方法无法实现编辑型和野生型的一次性检测,所以我们在传统PCR的基础上进行创新,即在引物5'端连接与本物种基因组序列不同源的功能核酸接头设计,这种设计可使野生型和编辑型等位基因扩增出大小不同且易于区分的产物;(3)扩增片段不宜过大,最好小于300 bp,以使相差几十bp的片段,通过琼脂糖凝胶电泳可以得到明显区分。
4 结论以MSTN编辑牛为研究材料,在传统PCR的基础上进行功能核酸接头设计的创新,建立的FNA-PCR方法不仅能够快速地检测出基因编辑动物,还能对其进行分型。本方法简便、灵敏度高、成本低,为基因编辑动物的检测提供了新思路。
| [1] |
Urnov F D, Miller J C, Lee Y L, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases[J]. Nat, 2005, 435(7042): 646-651. DOI:10.1038/nature03556 |
| [2] |
Boch J, Scholze H, Schornack S, et al. Breaking the code of DNA binding specificity of TAL-type Ⅲ effectors[J]. Science, 2010, 326(5): 1509-1512. |
| [3] |
Doudna J A, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9[J]. Science, 2015, 346(6213): 1258096. |
| [4] |
Cong L, Ran F A, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems[J]. Science, 2013, 8(11): 819-823. |
| [5] |
Lee S J, Mcpherron A C. Regulation of myostatin activity and muscle growth[J]. Proc Natl Acad Sci USA, 2001, 98(16): 9306-9311. DOI:10.1073/pnas.151270098 |
| [6] |
Mcpherron A C, Lee S J. Double muscling in cattle due to mutations in the myostatin gene[J]. Proc Natl Acad Sci USA, 1997, 94(23): 12457-12461. DOI:10.1073/pnas.94.23.12457 |
| [7] |
Rao S, Fujimura T, Matsunari H, et al. Efficient modification of the myostatin gene in porcine somatic cells and generation of knockout piglets[J]. Mol Reprod Dev, 2015, 83(1): 61-70. |
| [8] |
Hanset R, Michaux C, Dessy-Doize C, et al. Studies on the 7th rib cut in double muscled and conventional cattle. Anatomical, Histological and Biochemical Aspects[M]// Muscle Hypertrophy of Genetic Origin and its use to Improve Beef Production. Springer Netherlands, 1982: 341-349. https://link.springer.com/chapter/10.1007%2F978-94-009-7550-7_30
|
| [9] |
Shan Q, Wang Y, Li J, et al. Genome editing in rice and wheat using the CRISPR/Cas system[J]. Nat Protoc, 2014, 9(10): 2395. DOI:10.1038/nprot.2014.157 |
| [10] |
Mashal R D, Koontz J, Sklar J. Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases[J]. Nature Genetics, 1995, 9(2): 177-83. DOI:10.1038/ng0295-177 |
| [11] |
Wittwer CT, Reed GH, Gundry CN, et al. High-resolution genotyping by amplicon melting analysis using LCGreen[J]. Clin Chem, 2003, 49(6): 853-860. DOI:10.1373/49.6.853 |
| [12] |
Stoltenburg R, Reinemann C, Strehlitz B. SELEX—A(r)evolutionary method to generate high-affinity nucleic acid ligands[J]. Biomol Eng, 2007, 24(4): 381-403. DOI:10.1016/j.bioeng.2007.06.001 |
| [13] |
Silverman SK. Catalytic DNA:scope, applications, and biochemistry of deoxyribozymes[J]. Trends in Biochemical Sciences, 2016, 41(7): 595-609. DOI:10.1016/j.tibs.2016.04.010 |
| [14] |
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning:a laboratory manual[M]. 2nd ed. New York: Cold Spring Harbor Laboratory Press, 1989.
|
| [15] |
Yang P, Wang J, Gong G, et al. Cattle mammary bioreactor generated by a novel procedure of transgenic cloning for large-Scale production of functional human lactoferrin[J]. PLoS One, 2008, 3(10): e3453. DOI:10.1371/journal.pone.0003453 |
| [16] |
Maga E A, Cullor J S, Smith W, et al. Human lysozyme expressed in the mammary gland of transgenic dairy goats can inhibit the growth of bacteria that cause mastitis and the cold-spoilage of milk[J]. Foodborne Pathogens & Disease, 2006, 3(4): 384. |
| [17] |
Wang J, Yang P, et al. Expression and characterization of bioactive recombinant human alpha-lactalbumin in the milk of transgenic cloned cows[J]. J Dairy Sci, 2008, 91(12): 4466-4476. DOI:10.3168/jds.2008-1189 |
| [18] |
Hua Y, Wang C, Huang J, et al. A simple and efficient method for CRISPR/Cas9-induced mutant screening[J]. Journal of Genetics and Genomics, 2017, 44(4): 207-213. DOI:10.1016/j.jgg.2017.03.005 |
| [19] |
Vouillot L, Thélie A, Pollet N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by Engine-ered Nucleases[J]. G3(Bethesda), 2015, 5(3): 407-415. |
| [20] |
Thomas HR, Percival SM, Yoder BK, et al. High-throughput genome editing and phenotyping facilitated by high resolution melting curve analysis[J]. PLoS One, 2014, 9(12): e114632. DOI:10.1371/journal.pone.0114632 |