




2. 华中农业大学生命科学技术学院,武汉 430070;
3. 华中农业大学生物医学中心,武汉 430070
2. College of Life Science and Technology, Huazhong Agricultural University, Wuhan 430070;
3. Bio-Medical Center, Huazhong Agricultural University, Wuhani 430070
病原菌和宿主细胞通过长期的共同进化,许多革兰氏阴性菌,如肠致病性大肠杆菌(Enteropatho-genic Escherichia coli,EPEC)、肠出血性大肠杆菌(Enterohemorrhagic Escherichia coli,EHEC)、耶尔森菌(Yersinia)、志贺菌(Shigella)、沙门菌(Salmo-nella)、铜绿假单胞菌(Pseudomonas aeruginosa)已经进化出了复杂的能够将细菌编码的蛋白转运到真核细胞中去的三型分泌系统(Type Ⅲ secretion system,T3SS)。T3SS对病原菌的致病性至关重要,通过T3SS进入到宿主细胞中去的效应蛋白能够通过精细的调控,来操纵细胞内的生物学过程,从而有效的逃逸宿主细胞的免疫清除反应。其中,细胞凋亡和细胞焦亡Pryoptosis是宿主细胞清除致病菌、维持机体稳态的两种主要途径,也是以上几种革兰氏阴性菌的T3SS效应蛋白操纵宿主细胞死亡的两个主要方面,但是不同的效应蛋白在该过程中发挥的功能相似或相反。本文就目前已发现的T3SS效应蛋白干扰细胞凋亡和焦亡信号通路的研究进展进行综述,并进行全面归纳比较和分析,揭示这些细菌分泌的效应蛋白的生物学功能,以进一步推动病原菌T3SS效应蛋白尚未发现的功能、生理底物和致病机制的研究,旨在为药物开发提供策略和靶点,以及为病原菌的预防和治疗提供理论依据。
1 对细胞凋亡的调控细胞凋亡(Apoptosis)指为维持内环境稳定,由基因控制的细胞自主的有序的死亡。细胞凋亡引起的程序性细胞死亡是一个严格的调控过程,由内源性(内质网和线粒体介导的)或外源性(受体介导的)途径激活,均导致caspase级联反应的激活,最终导致细胞皱缩,染色质凝聚,细胞膜出泡及小泡芽形成,即形成凋亡小体[1]。细胞凋亡是宿主抵抗细胞内病原的最后防御策略,即通过引起细胞程序性死亡,宿主可阻止入侵微生物在细胞内的增殖[2]。病原菌已经进化出通过注入特定的T3SS效应蛋白来操纵宿主细胞死亡的防御机制。
1.1 抑制细胞凋亡感染了细菌的宿主细胞可通过凋亡的方式防止感染的扩散。某些T3SS效应蛋白干扰上皮细胞凋亡的起始阶段来促进形成一个合适的生存环境。对于某些肠道致病菌而言,抑制宿主细胞的程序性死亡,一方面有利于细菌在肠道表面的附着和增殖;另一方面则可以限制炎症反应的扩大,帮助细菌逃逸宿主免疫系统的攻击[3]。
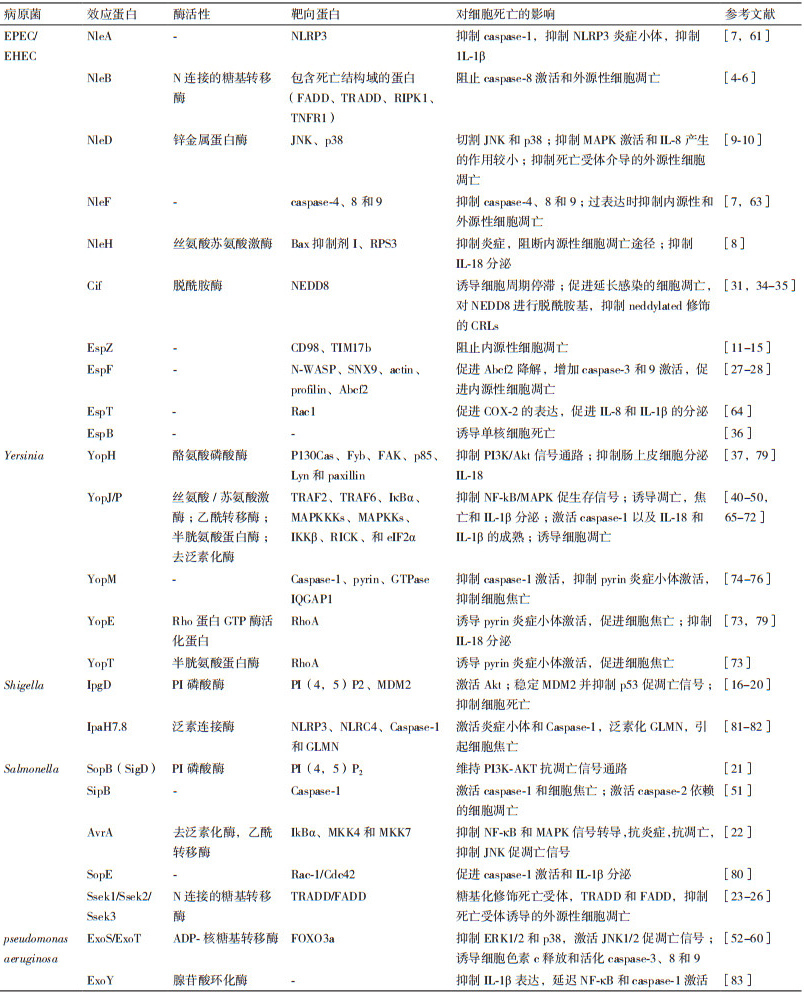
EPEC/EHEC通过多种T3SS效应蛋白来操纵宿主细胞的细胞凋亡传导通路。我们前期的研究以及Li和Pearson等[4-5]的研究表明,NleB是一种糖基转移酶,特异性地对TRADD、FADD和RIPK1等多个接头蛋白死亡结构域中的一个保守精氨酸进行N-乙酰葡萄糖胺化修饰,阻止死亡受体复合物的形成和caspase-8的切割,从而抑制TNFα、FasL和TRAIL等死亡受体配体诱导宿主细胞发生凋亡和坏死,Scott等[6]的进一步研究表明,NleB优先修饰FADD 117位精氨酸,从而阻断死亡受体诱导的细胞死亡。而瞬时表达EPEC/EHEC的效应蛋白NleF时,NleF可以特异性结合并抑制caspase-4、8和9的活性,抑制十字孢碱和TRAIL诱导的细胞死亡[7]。EPEC/EHEC的效应蛋白NleH家族可以阻断由十字孢碱、布雷菲德菌素A和衣霉素刺激的细胞凋亡,后两者诱导内源性细胞凋亡途径,导致Bax构象改变并转位到线粒体,线粒体发生透化,细胞色素c释放。NleH通过和促凋亡蛋白Bax抑制因子-Ⅰ(BI-Ⅰ)直接的相互作用,阻断内源性途径,然而BI-1的作用机理并不明确[8]。此外,EPEC的效应蛋白NleD通过切割JNK的活性也参与到细胞凋亡的抑制。在感染期间,NleD异位表达时抑制UV诱导的凋亡(JNK依赖性的)[9]。进一步的研究表明,NleD在JNK激酶激活环的TPY基序Y185之前进行切割,在p38的M213位点进行切割,并且保持NleD酶活性的R203位点在切割p38中起到关键作用,从而抑制细胞凋亡[10]。
效应蛋白EspZ抑制感染上皮细胞诱导的细胞死亡[11-12]。异位表达EspZ抑制十字孢碱诱导的细胞死亡[12]。EspZ作用的机制最初被报道为参与和宿主细胞蛋白CD98的相互作用,可以增强β1-整合素下游的促生存信号[13]。然而,另一个研究表明,在小肠上皮细胞中,该功能的作用机制对于EspZ的抗凋亡作用不是必需的[12]。EspZ的抗凋亡功能也是由于和线粒体内膜转位酶17b(TIM17b)的相互作用[14],2014有研究表明,EspZ通过阻止EspF诱导的表皮生长因子(EGFR)的缺失避免细胞死亡[15]。而另一个模型提示EspZ通过控制T3SS效应蛋白转位维持细胞存活,可能是阻止促凋亡效应蛋白转位到细胞[11]。因此,EspZ阻止感染的细胞凋亡过程中的作用机制有待进一步研究。总之,效应蛋白NleB、NleF、NleH、NleD、EspZ通过抑制细胞凋亡途径或者增强促生存信号来阻止细胞死亡。
在Shigella感染后,效应蛋白IpgD诱导早期活化的磷酸化Mdm2蛋白的形成,进而促进p53的降解,同时,效应蛋白VirA激活钙蛋白酶,也可以促进p53的降解,而p53的降解阻断了NF-κB依赖的促凋亡信号通路[16]。另一个研究表明效应蛋白IpgD通过PtdIns(5)P激活PI3K/Akt[17],从而进一步促进磷酸化Mdm2蛋白的激活[18],导致细胞凋亡的抑制。Pendaries等[17]的研究也证明了IpgD可以抑制十字孢碱诱导的HEK293细胞凋亡,这与Clark等[19]证明的IpgD对于抑制十字孢碱诱导的HeLa细胞凋亡并不是必需的结论相反,这可能是由于处理的细胞和时间不同引起的,而IpgD对不同细胞凋亡的影响还有待进一步研究。最新研究发现,Shigella的效应蛋白OspD2和IpaH1.4是细胞死亡抑制剂。与大多数T3SS相反,OspD2并不靶向细胞过程,而是调节Shigella三型分泌装置的活性,抑制效应蛋白VirA诱导的钙蛋白酶介导的坏死性上皮细胞死亡[20]。
Salmonella通过效应蛋白SopB、AvrA和SseKs促进生存并抑制凋亡来影响宿主细胞凋亡。SopB具有磷酸肌醇磷酸酶活性,在鼠伤寒沙门菌(S. enterica serovar Typhimurium)感染的上皮细胞中,以SopB依赖的方式诱导促生存Akt/蛋白激酶B持续的激活,抑制喜树碱诱导的Caspase-3切割和细胞凋亡[21]。AvrA通过它的乙酰转移酶活性在丝裂原活化蛋白激酶4和7(MKK4和MKK7)水平抑制c-Jun NH2-末端激酶(JNK)信号通路激活,抑制Salmonella感染上皮细胞引起的先天性免疫和细胞凋亡[22]。另外,Salmonella的效应蛋白SseK1、SseK2和SseK3与NleB同源,研究显示SseK1和SseK3具有N-乙酰糖基转移酶活性,SseK1糖基化修饰TRADD和FADD,SseK3修饰FADD,两者均抑制死亡受体诱导的细胞凋亡[23];而El Qaidi等[24]的研究表明,SseK1糖基化修饰GAPDH,SseK2修饰FADD,而SseK3既不修饰GAPDH也不修饰FADD,结果表明SseK1、SseK和SseK3分别靶向不同的宿主底物蛋白。Esposito等[25]的研究进一步说明了SseK3具有糖基转移酶活性,可以糖基化修饰TRADD。最新的结构研究揭示了SseK1和SseK2结合底物时的结构不同[26]。
1.2 诱导细胞凋亡一方面病原菌可以分泌T3SS效应蛋白阻断细胞凋亡来维持它们在宿主细胞内的黏附和增殖;另一方面,病原菌T3SS的一些效应蛋白可作用于特定的信号转导通路,诱导宿主细胞的程序性死亡来扩大感染。
在EPEC/EHEC感染期间,T3SS效应蛋白包括EspF、Cif、EspH和EspB激活内源性细胞死亡。当EPEC感染HeLa细胞时,EspF靶向线粒体,引起线粒体外膜电位的损失,细胞色素C释放,以及Caspase-9和Caspase-3的切割,因此EspF在起始线粒体死亡信号通路中发挥作用[27]。另一项研究表明,EspF结合ABC转运蛋白家族的Abcf2,在感染期间,Abcf2以EspF依赖的方式降解,导致caspase-9和caspase-3的切割增加,促进细胞凋亡[28]。EPEC/EHEC的效应蛋白Cif阻断细胞周期进程并且最近被鉴定为EPEC诱导的细胞凋亡的晚期诱导物,可以引起caspase-3切割和乳酸脱氢酶(LDH)释放[29]。Cif在细胞凋亡中的作用可能和它抑制泛素/蛋白酶体途径的功能有关。在EPEC感染期间,Cif引起细胞病变效果(CPE),特征为逐渐募集黏着斑,引起应力纤维的组装并最终抑制细胞周期的G1/S期转变,引起延迟的细胞死亡[30-32]。Cif阻断宿主细胞周期进程的机制为,cullin-RING泛素连接酶(CRLs)调节几种参与细胞周期进程的蛋白质,类泛素蛋白NEDD8对CRLs进行neddylation修饰后,CRLs活性大大增加[33]。Cif具有脱氨酶活性,能够催化NEDD8脱氨,脱氨的NEDD8对CRLs进行neddylation修饰后,CRLs的活性大大减弱,进而抑制细胞周期进程[31, 34-35]。最新发现,EPEC的T3SS效应蛋白EspB可以自主进入宿主细胞,并选择性诱导单核细胞程序性细胞死亡[36]。
在Y.pestis中,YopH是一种酪氨酸磷酸酶,可以激活Caspases,引起线粒体膜电势损失,诱导线粒体介导的T细胞凋亡[37],而YpkA和YopM激活上皮细胞和/或巨噬细胞中的caspase-3[38-39]。Yersinia的YopP/YopJ在未活化的巨噬细胞中激活caspase-8[40]。大量研究表明,Y. enterocolitica的效应蛋白YopP与Y. pseudotuberculosis中的YopJ相类似,在感染引起的巨噬细胞凋亡过程中发挥重要作用[41-42]。YopJ是去泛素半胱氨酸蛋白酶[43-44],同时也具有乙酰转移酶的功能,可以乙酰化MEK2[45-46],能有效抑制促生存MAPK和NF-κB信号通路[47],也可以通过TLR4信号通路促进细胞凋亡[48]。而YopP可裂解caspase-8的底物BID来促进细胞凋亡[49-50]。
Salmonella诱导细胞凋亡的研究较少,研究显示,Salmonella以SipB依赖的方式感染巨噬细胞诱导caspase-2切割,导致不依赖于caspase-1的细胞凋亡和细胞死亡[51]。
P. aeruginosa的T3SS效应蛋白ExoS和ExoT都具有GTP酶(GAP)和ADP核糖基转移酶(ADPRT)结构[52-53],ExoS和ExoT均能诱导细胞凋亡[54]。ExoS的ADP-核糖基转移酶活性对诱导细胞凋亡是必须的[55]。在P. aeruginosa诱导细胞凋亡过程中ERK1/2和p38等宿主细胞存活信号通路的活化被抑制,而JNK1/2促凋亡信号被激活[56]。并且ExoS可抑制FOXO3a的磷酸化,并诱导细胞色素c释放和活化caspase-3、8和9,从而诱导细胞凋亡[57-58]。另一项研究表明ExoS的GAP活性是通过靶向维持宿主细胞肌动蛋白骨架的小GTP酶,诱导细胞分离,抑制细胞迁移和吞噬作用。其ADPRT结构域是通过抑制宿主细胞DNA合成,改变内吞作用,引起细胞凋亡,从而有利于P. aeruginosa在细胞内繁殖[53]。ExoT的GAP活性通过激活JNK1/2、大幅度增加线粒体的促凋亡蛋白Bax和Bid产生、促进细胞色素c的释放以及激活caspase-9和caspase-3,引起线粒体途径的细胞凋亡,ExoT的ADPRT结构域通过改变一种细胞蛋白Crk产生细胞毒素,干扰整合素的生存信号,来诱导细胞凋亡[59]。最新研究证明ExoS的GAP结构域是必需的并且足以诱导线粒体途径的细胞凋亡。效应蛋白ExoS的GAP结构域导致Bax和Bim富集到线粒体外膜,破坏线粒体膜并释放细胞色素c到细胞质,进而激活caspase-9和caspase-3,从而导致细包凋亡[60]。
2 对细胞焦亡的调控细胞焦亡(Pyroptosis)是近年来新发现的一种炎症性程序性细胞死亡,形态学上表现为细胞DNA断裂、细胞质空泡化和细胞膜破损。细胞质感应器如Nod-like受体(NLRs,包括NLRP3和NLRC4)、pyrin、caspases-4和5感应到细菌组分后介导炎症小体的组装,随后激活caspase-1导致IL-1β和IL-18前体切割成它们的激活形式。炎症小体在宿主抵抗感染中发挥关键作用,但是,炎症小体产生过多,存在时间过长,炎症和细胞因子的过量表达和持续作用,则可造成细胞焦亡,进而引起慢性炎症、组织损伤和细胞死亡。细胞焦亡的重要作用是从细胞中释放游离的细菌,这个过程有利于细菌的传播,而宿主细胞死亡却有利于促进宿主细胞清除细菌。
EPEC和EHEC通过许多效应蛋白来干扰炎症反应,从而潜在地影响细胞焦亡。NleA通过和PYD和LRR结构域相互作用直接靶向NLRP3,阻止NLRP3的去泛素,抑制NLRP3炎症小体形成,并且NleA抑制caspase-1激活,进而抑制宿主分泌IL-1β[61]。NleF是几个Caspase家族成员的抑制剂,包括Caspases-4、8和9[7],由于Caspase-4和Caspase-8都明确参与到非典型NLRP3炎症小体激活[62],因此,EPEC/EHEC可能通过抑制caspase-4和caspase-8潜在地干扰NLRP3炎症小体[7]。最新研究表明,NleF通过抑制caspase-4-p19和caspase-4-p10相互作用以及抑制caspase-1和caspase-4相互作用,进而抑制细胞焦亡和IL-1β产生[63]。EspT诱导促进炎症的环氧化酶-2(COX-2)的表达,并且发现Rac1参与到EspT诱导的IL-8和IL-1β的分泌[64]。因此NleA、NleF和EspT通过干扰炎症反应相关成分,潜在地影响细胞焦亡。
Yersinia感染的细胞也观察到焦亡的特点。Yersinia主要通过干扰宿主caspase-1以及NLRP3、NLRC4和Pyrin炎症小体来抵抗宿主应答。Yersinia通过激活caspase-1诱导巨噬细胞死亡。效应蛋白YopJ引起巨噬细胞中促凋亡蛋白酶caspase-3的激活被显著地延迟并导致caspase-1依赖的细胞焦亡[65]。Zheng等[66]的实验表明,原代小鼠巨噬细胞中YopJ可以诱导细胞死亡,caspase-1激活,和IL-1β分泌。Philip等[67]的研究表明,YopJ诱导的细胞死亡和caspase-1激活需要RIPK1,FADD以及caspase-8。而Ratner和Schoberle[68-69]发现YopJ和YopM一起发挥作用来阻止炎症小体激活。YopJ由于具有干扰MyD88和Trif依赖的应答的产生的能力而限制炎症小体引发[70],分泌效应蛋白YopJ引起caspase-8依赖的IL-1β激活。T3SS的针/转位成分激活NLRP3和NLRC4依赖的IL-1β产生[71]。发现体外实验中,Y. pestis的效应蛋白YopJ单独可以引起巨噬细胞死亡,抑制炎性细胞聚集到深部组织中的细菌生长处[72]。效应蛋白YopE是一个GTPase激活蛋白,通过促进GTP水解模拟真核GTPase激活蛋白GAPs。YopT是切割Rho-GTPase的半胱氨酸蛋白酶。研究表明,YopE和YopT均可靶向并失活RhoA,导致pyrin炎症小体的激活[73]。与效应蛋白YopJ、YopE和YopT相反,YopM、YopK、YopB和YopD抑制炎症小体激活。YopM可以直接结合并抑制caspase-1活性,阻断炎症小体形成,进而阻止焦亡性细胞死亡来应答Yersinia感染[74],也可以与GTPase IQGAP1相互作用,限制炎症小体激活[73],同时,YopM也通过靶向pyrin炎症小体限制caspase-1激活[75]。研究表明,YopM抑制caspase-1依赖的细胞焦亡[68],阻止YopE诱导的Pyrin炎症小体的激活,阻断Pyrin介导的caspase-1依赖的IL-1β/IL-18产生和细胞死亡[71]。YopM结合并激活PRK和RSK[75],YopM/PRK/RSK复合物能磷酸化pyrin,磷酸化的pyrin结合到14-3-3蛋白,而14-3-3蛋白抑制pyrin炎症小体,因此YopM抑制pyrin炎症小体[76],从而促进Yersinia在体内的毒力。Yersinia效应蛋白YopK和T3SS转位装置的成分YopB和YopD相互作用,限制它们异常转位到感染的宿主细胞,从而限制NLRP3和NLRC4炎症小体的激活[77-78]。Y. enterocolitica的YopE和YopH抑制肠上皮细胞(IECs)分泌激活的IL-18,IL-18分泌依赖caspase-1和NLRP3炎症小体[79]。
Salmonella有关此方面的报道比较少,其中S. Typhimurium的T3SS效应蛋白SopE,是宿主细胞内Rho GTPases的激活剂,通过Rho GTPases Rac-1和Cdc42导致caspase-1激活以及随后的细胞因子IL-1β成熟和分泌[80]。
研究显示,Shigella通过T3SS分泌IpaH7.8 E3泛素连接酶效应蛋白,从而激活NLRP3和NLRC4炎症小体,以及caspase-1并且IpaH7.8泛素化肾小球蛋白(GLMN)而引起GLMN的降解,从而引起炎症小体激活和巨噬细胞焦亡[81-82]。最新研究显示,P. aeruginosa的效应蛋白ExoY具有腺苷酸环化酶活性,ExoY可以抑制IL-1β表达,延迟NF-κB和caspase-1激活[83]。
总之,EPEC/ EHEC抑制细胞焦亡并控制上皮细胞凋亡来保证其合适的生存位置。而Yersinia通过抑制巨噬细胞焦亡来避免引起炎症反应,促进巨噬细胞凋亡以维持细胞外存活。Shigella和Salmonella引发巨噬细胞焦亡来促进炎症反应,这有助于有效地侵袭肠道屏障。
本文中主要的三型分泌系统效应蛋白对细胞凋亡和焦亡的影响总结,见表 1。
三型分泌系统效应蛋白对宿主细胞的影响近年来一直是研究的热点和难点。病原菌感染后或者被宿主的天然免疫系统清除,或者能够逃避宿主的免疫防御机制而导致宿主发病。病原菌逃避宿主先天性免疫的机制有多种,其中包括对宿主细胞死亡的影响。这里我们回顾了肠致病性大肠杆菌/肠出血性大肠杆菌,耶尔森菌,沙门菌,志贺菌和铜绿假单胞菌的T3SS效应蛋白在影响细胞凋亡和细胞焦亡过程中的作用,发现不同病原菌的效应蛋白具有不同或者相似的功能,而同一种病原菌的效应蛋白之间具有协同或拮抗作用,通过操纵细胞凋亡或者细胞焦亡信号通路来促进或者抑制宿主细胞死亡,进而促进病原菌的增殖和传播。然而,关于病原菌T3SS效应蛋白发挥作用的机制仍有许多需要了解,一些效应蛋白的未知生理底物及该底物的功能需要深入研究。研究效应蛋白如何促进或阻遏甚至中断宿主细胞凋亡或者焦亡的过程,不仅可以帮助我们深入理解病原菌的致病机制,更为防治具有T3SS病原菌药物的开发提供新思路和新靶点。
由于抗菌药物的滥用使致病菌受到强烈的生存压力进而导致耐药菌株的产生,因此既要采取各种措施减少菌株耐药性的发展和传播,又要寻找既不会影响病原体自身生长又可以有效降低其致病性的抗毒力药物。而T3SS是存在于革兰氏阴性致病菌中高度保守的毒力因子[84],研究表明T3SS抑制剂在抑制毒性系统时,对细菌的生长几乎没有影响,这意味着不会产生细菌耐药性问题[85]。这使得T3SS正成为非常有吸引力的新颖抗菌药物的重要靶标[83]。近年来研究者利用各种筛选系统从化合物库和天然产物库中筛选得到不同的T3SS抑制剂[86-87],其作用机制包括抑制T3SS效应蛋白的分泌[86],如亚水杨基酰肼类衍生物INP0341和羟基喹啉类IP1750抑制ExoS的分泌[88];苯氧乙酰胺类化合物MBX1641可以抑制ExoS和ExoT蛋白的表达[87];甘草黄酮醇(Licoflavonol,LCF)可以抑制S. enterica serovar Typhimurium的T3SS效应蛋白SipC分泌[89]。但针对大多数抑制剂,其作用的真正靶点尚未找到,鉴于T3SS效应蛋白对宿主细胞的凋亡和焦亡影响在致病过程中发挥了重要作用,因此对效应蛋白作用机制的进一步研究将会对T3SS抑制剂的开发有巨大的促进作用,为开发防治病原菌新靶点的抑制剂提供思路。
| [1] |
Taylor RC, Cullen SP, Martin SJ. Apoptosis:controlled demolition at the cellular level[J]. Nat Rev Mol Cell Biol, 2008, 9(3): 231-241. DOI:10.1038/nrm2312 |
| [2] |
Faherty CS, Maurelli AT. Staying alive:bacterial inhibition of apoptosis during infection[J]. Trends Microbiol, 2008, 16(4): 173-180. DOI:10.1016/j.tim.2008.02.001 |
| [3] |
Giogha C, Lung TW, Pearson JS, et al. Inhibition of death receptor signaling by bacterial gut pathogens[J]. Cytokine Growth Factor Rev, 2014, 25(2): 235-243. DOI:10.1016/j.cytogfr.2013.12.012 |
| [4] |
Li S, Zhang L, Yao Q, et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains[J]. Nature, 2013, 501(7466): 242-246. DOI:10.1038/nature12436 |
| [5] |
Pearson JS, Giogha C, Ong SY, et al. A type Ⅲ effector antagonizes death receptor signalling during bacterial gut infection[J]. Nature, 2013, 501(7466): 247-251. DOI:10.1038/nature12524 |
| [6] |
Scott NE, Giogha C, Pollock GL, et al. The bacterial arginine glycosyltransferase effector NleB preferentially modifies Fas-associated death domain protein(FADD)[J]. J Biol Chem, 2017, 292(42): 17337-17350. DOI:10.1074/jbc.M117.805036 |
| [7] |
Blasche S, Mörtl M, Steuber H, et al. The E. coli effector protein NleF is a caspase inhibitor[J]. PLoS One, 2013, 8(3): e58937. DOI:10.1371/journal.pone.0058937 |
| [8] |
Hemrajani C, Berger CN, Robinson KS, et al. NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection[J]. Proc Natl Acad Sci USA, 2010, 107(7): 3129-3134. DOI:10.1073/pnas.0911609106 |
| [9] |
Baruch K, et al. Metalloprotease type Ⅲ effectors that specifically cleave JNK and NF-κB[J]. EMBO J, 2011, 30(1): 221-231. DOI:10.1038/emboj.2010.297 |
| [10] |
Creuzburg K, et al. The type Ⅲ effector NleD from enteropathogenic Escherichia coli differentiates between host substrates p38 and JNK[J]. Infect Immun, 2017, 85(2). pii: e00620-e00616. https://www.ncbi.nlm.nih.gov/pubmed/27872241
|
| [11] |
Berger CN, et al. EspZ of enteropathogenic and enterohemorrhagic Escherichia coli regulates type Ⅲ secretionsystem protein translocation[J]. MBio, 2012, 3(5), pii: e00317-e00312. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM23033475
|
| [12] |
Roxas JL, Wilbur JS, Zhang X, et al. The enteropathogenic Escherichia coli-secreted protein EspZ inhibits host cell apoptosis[J]. Infect Immun, 2012, 80(11): 3850-3857. DOI:10.1128/IAI.00335-12 |
| [13] |
Shames SR, et al. The pathogenic E. coli type Ⅲ effector EspZ interacts with host CD98 and facilitates host cell prosurvival signaling[J]. Cell Microbiol, 2010, 12(9): 1322-1339. DOI:10.1111/j.1462-5822.2010.01470.x |
| [14] |
Shames SR, Croxen MA, Deng W, et al. The type Ⅲ system-secreted effector EspZ localizes to host mitochondria and interacts with the translocase of inner mitochondrial membrane 17b[J]. Infect Immun, 2011, 79(12): 4784-4790. DOI:10.1128/IAI.05761-11 |
| [15] |
Roxas JL, et al. Enteropathogenic Escherichia coli dynamically regulates EGFR signaling in intestinal epithelial cells[J]. Am J Physiol Gastrointest Liver Physiol, 2014, 307(3): G374-380. DOI:10.1152/ajpgi.00312.2013 |
| [16] |
Bergounioux J, Elisee R, Prunier AL, et al. Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium's epithelial niche[J]. Cell Host Microbe, 2012, 11(3): 240-252. DOI:10.1016/j.chom.2012.01.013 |
| [17] |
Pendaries C, Tronchère H, Arbibe L, et al. PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection[J]. EMBO J, 2006, 25(5): 1024-1034. DOI:10.1038/sj.emboj.7601001 |
| [18] |
Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from thecytoplasm to the nucleus[J]. Proc Natl Acad Sci USA, 2001, 98(20): 11598-11603. DOI:10.1073/pnas.181181198 |
| [19] |
Clark CS, Maurelli AT. Shigella flexneri inhibits staurosporine-induced apoptosis in epithelial cells[J]. Infect Immun, 2007, 75(5): 2531-2539. DOI:10.1128/IAI.01866-06 |
| [20] |
Mou X, et al. Synthetic bottom-up approach reveals the complex interplay of Shigella effectors in regulation of epithelial cell death[J]. Proc Natl Acad Sci USA, 2018, 115(25): 6452-6457. DOI:10.1073/pnas.1801310115 |
| [21] |
Knodler LA, Finlay BB, Steele-Mortimer O. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt[J]. J Biol Chem, 2005, 10: 9058-9064. |
| [22] |
Jones RM, Wu H, et al. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade[J]. Cell Host Microbe, 2008, 3(4): 233-244. DOI:10.1016/j.chom.2008.02.016 |
| [23] |
Günster RA, Matthews SA, Holden DW, et al. SseK1 and SseK3 type Ⅲ secretion system effectors inhibit NF-κB signaling and necroptotic cell death in Salmonella-Infected Macrophages[J]. Infect Immun. 2017, 85(3), pii: e00010- e00017.
|
| [24] |
El Qaidi S, Chen K, Halim A, et al. NleB/SseK effectors from Citrobacter rodentium, Escherichia coli, and Salmonella enterica display distinct differences in host substrate specificity[J]. J Biol Chem, 2017, 292(27): 11423-11430. DOI:10.1074/jbc.M117.790675 |
| [25] |
Esposito D, Günster RA, Martino L, et al. Structural basis for the glycosyltransferase activity of the Salmonella effector SseK3[J]. J Biol Chem, 2018, 293(14): 5064-5078. DOI:10.1074/jbc.RA118.001796 |
| [26] |
Park JB, Kim YH, Yoo Y, et al. Structural basis for arginine glycosylation of host substrates by bacterial effector proteins[J]. Nat Commun, 2018, 9(1): 4283. DOI:10.1038/s41467-018-06680-6 |
| [27] |
Nougayrède JP, Donnenberg MS. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway[J]. Cell Microbiol, 2004, 6(11): 1097-1111. DOI:10.1111/cmi.2004.6.issue-11 |
| [28] |
Nougayrède JP, Foster GH, Donnenberg MS. Enteropathogenic Escherichia coli effector EspF interacts with host protein Abcf2[J]. Cell Microbiol, 2007, 9(3): 680-693. DOI:10.1111/cmi.2007.9.issue-3 |
| [29] |
Samba-Louaka A, Nougayrède JP, Watrin C, et al. The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells[J]. Infect Immun, 2009, 77(12): 5471-5477. DOI:10.1128/IAI.00860-09 |
| [30] |
De Rycke J, Comtet E, Chalareng C, et al. Enteropathogenic Escherichia coli O103 from rabbit elicits actin stress fibers and focal adhesions in HeLa epithelial cells, cytopathic effects that are linked to an analog of the locus of enterocyte effacement[J]. Infect Immun, 1997, 65(7): 2555-2563. |
| [31] |
Morikawa H, et al. The bacterial effector Cif interferes with SCF ubiquitin ligase function by inhibiting deneddylationof Cullin1[J]. Biochem Biophys Res Commun, 2010, 2: 268-274. |
| [32] |
Nougayrède JP, et al. Type Ⅲ secretion-dependent cell cycle block caused in HeLa cells by enteropathogenic Escherichia coli O103[J]. Infect Immun, 2001, 11: 6785-6795. |
| [33] |
Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases[J]. Nat Rev Mol Cell Biol, 2005, 6(1): 9-20. |
| [34] |
Cui J, Yao Q, Li S, et al. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family[J]. Science, 2010, 329(5996): 1215-1218. DOI:10.1126/science.1193844 |
| [35] |
Toro TB, Toth JI, Petroski MD. The cyclomodulin cycle inhibiting factor(CIF)alters cullin neddylation dynamics[J]. J Biol Chem, 2013, 288(21): 14716-14726. DOI:10.1074/jbc.M112.448258 |
| [36] |
Baumann D, et al. Multitalented EspB of enteropathogenic Escherichia coli(EPEC)enters cells autonomously and induces programmed cell death in human monocytic THP-1 cells[J]. Int J Med Microbiol, 2018, 308(3): 387-404. DOI:10.1016/j.ijmm.2018.03.005 |
| [37] |
Bruckner S, Rhamouni S, Tautz L, et al. Yersinia phosphatase induces mitochondrially dependent apoptosis of T cells[J]. J Biol Chem, 2005, 280(11): 10388-10394. DOI:10.1074/jbc.M408829200 |
| [38] |
Pha K, Navarro L. Yersinia type Ⅲ effectors perturb host innate immune responses[J]. World J Biol Chem, 2016, 7(1): 1-13. DOI:10.4331/wjbc.v7.i1.1 |
| [39] |
Ye Z, Gorman AA, Uittenbogaard AM, et al. Caspase-3 mediates the pathogenic effect of Yersinia pestis YopM in liver of C57BL/6 mice and contributes to YopM's function in spleen[J]. PLoS One, 2014, 9(11): e110956. DOI:10.1371/journal.pone.0110956 |
| [40] |
Philip NH, Zwack EE, Brodsky IE. Activation and Evasion of Inflammasomes by Yersinia[J]. Curr Top Microbiol Immunol, 2016, 397: 69-90. |
| [41] |
Mills SD, Boland A, Sory MP, et al. Yersinia enterocolitica induces apoptosis in macrophages by a process requiring functional type Ⅲ secretion and translocation mechanisms and involving YopP, presumably acting as an effector protein[J]. Proc Natl Acad Sci USA, 1997, 94(23): 12638-12643. DOI:10.1073/pnas.94.23.12638 |
| [42] |
Monack DM, Mecsas J, Ghori N, et al. Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death[J]. Proc Natl Acad Sci USA, 1997, 94(19): 10385-10390. DOI:10.1073/pnas.94.19.10385 |
| [43] |
Orth K, Xu Z, Mudgett MB, et al. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease[J]. Science, 2000, 290(5496): 1594-1597. DOI:10.1126/science.290.5496.1594 |
| [44] |
Zhou H, Monack DM, Kayagaki N, et al. Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-kappa B activation[J]. J Exp Med, 2005, 202(10): 1327-1332. DOI:10.1084/jem.20051194 |
| [45] |
Mittal R, Peak-Chew SY, McMahon HT. Acetylation of MEK2 and I kappa B kinase(IKK)activation loop residues by YopJ inhibits signaling[J]. Proc Natl Acad Sci USA, 2006, 103(49): 18574-18579. DOI:10.1073/pnas.0608995103 |
| [46] |
Mukherjee S, Keitany G, Li Y, et al. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation[J]. Science, 2006, 312(5777): 1211-1214. DOI:10.1126/science.1126867 |
| [47] |
Denecker G, et al. Yersinia enterocolitica YopP-induced apoptosis of macrophages involves the apoptotic signaling cascade upstream of bid[J]. J Biol Chem, 2001, 276(23): 19706-19714. DOI:10.1074/jbc.M101573200 |
| [48] |
Gröbner S, Adkins I, Schulz S, et al. Catalytically active Yersinia outer protein P induces cleavage of RIP and caspase-8 at the level of the DISC independently of death receptors in dendritic cells[J]. Apoptosis, 2007, 12(10): 1813-1825. DOI:10.1007/s10495-007-0100-x |
| [49] |
Viboud GI, Bliska JB. Yersinia outer proteins:role in modulation of host cell signaling responses and pathogenesis[J]. Annu Rev Microbiol, 2005, 59: 69-89. DOI:10.1146/annurev.micro.59.030804.121320 |
| [50] |
Zhang Y, Bliska JB. Role of Toll-like receptor signaling in the apoptotic response of macrophages to Yersinia infection[J]. Infect Immun, 2003, 71(3): 1513-1519. DOI:10.1128/IAI.71.3.1513-1519.2003 |
| [51] |
Jesenberger V, et al. Salmonella-induced caspase-2 activation in macrophages:a novel mechanism in pathogen-mediated apoptosis[J]. J Exp Med, 2000, 192(7): 1035-1046. DOI:10.1084/jem.192.7.1035 |
| [52] |
Kazmierczak BI, Engel JN. Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42[J]. Infect Immun, 2002, 70(4): 2198-2205. DOI:10.1128/IAI.70.4.2198-2205.2002 |
| [53] |
Heimer SR, et al. Pseudomonas aeruginosa utilizes the type Ⅲ secreted toxin ExoS to avoid acidified compartments within epithelial cells[J]. PLoS One, 2013, 8(9): e73111. DOI:10.1371/journal.pone.0073111 |
| [54] |
Hauser AR. The type Ⅲ secretion system of Pseudomonas aeruginosa:infection by injection[J]. Nat Rev Microbiol, 2009, 7(9): 654-665. DOI:10.1038/nrmicro2199 |
| [55] |
Kaufman MR, Jia J, Zeng L, et al. Pseudomonas aeruginosa mediated apoptosis requires the ADP-ribosylating activity of exoS[J]. Microbiology, 2000, 146(Pt 10): 2531-2541. |
| [56] |
Jia J, et al. c-Jun NH2-terminal kinase-mediated signaling is essential for Pseudomonas aeruginosa ExoS-induced apoptosis[J]. Infect Immun, 2003, 71(6): 3361-3370. DOI:10.1128/IAI.71.6.3361-3370.2003 |
| [57] |
Alaoui-El-Azher M, Jia J, Lian W, et al. ExoS of Pseudomonas aeruginosa induces apoptosis through a Fas receptor/caspase 8-independent pathway in HeLa cells[J]. Cell Microbiol, 2006, 8(2): 326-338. DOI:10.1111/cmi.2006.8.issue-2 |
| [58] |
Jansson AL, Yasmin L, Warne P, et al. Exoenzyme S of Pseudomonas aeruginosa is not able to induce apoptosis when cells express activated proteins, such as Ras or protein kinase B/Akt[J]. Cell Microbiol, 2006, 8(5): 815-822. DOI:10.1111/cmi.2006.8.issue-5 |
| [59] |
Wood SJ, et al. Pseudomonas aeruginosa ExoT induces mitochondrial apoptosis in target host cells in a manner that depends on its GTPase-activating protein(GAP)domain activity[J]. J Biol Chem, 2015, 290(48): 29063-29073. DOI:10.1074/jbc.M115.689950 |
| [60] |
Kaminski A, Gupta KH, Goldufsky JW, et al. Pseudomonas aeruginosa ExoS induces intrinsic apoptosis in target host cells in a manner that is dependent on its GAP domain activity[J]. Sci Rep, 2018, 8(1): 14047. DOI:10.1038/s41598-018-32491-2 |
| [61] |
Yen H, Sugimoto N, Tobe T. Enteropathogenic Escherichia coli uses NleA to inhibit NLRP3 inflammasome activation[J]. PLoS Pathog, 2015, 11(9): e1005121. DOI:10.1371/journal.ppat.1005121 |
| [62] |
Echtenkamp F, Deng W, Wickham ME, et al. Characterization of the NleF effector protein from attaching and effacing bacterial pathogens[J]. FEMS Microbiol Lett, 2008, 281(1): 98-107. DOI:10.1111/fml.2008.281.issue-1 |
| [63] |
Song T, Li K, Zhou W, et al. A Type Ⅲ effector NleF from EHEC inhibits epithelial inflammatory cell death by targeting caspase-4[J]. Biomed Res Int, 2017, 2017: 4101745. |
| [64] |
Raymond B, et al. The WxxxE effector EspT triggers expression of immune mediators in an Erk/JNK and NF-κB-dependent manner[J]. Cell Microbiol, 2011, 13(12): 1881-1893. DOI:10.1111/j.1462-5822.2011.01666.x |
| [65] |
Bergsbaken T, Cookson BT. Macrophage activation redirects Yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis[J]. PLoS Pathog, 2007, 3(11): e161. DOI:10.1371/journal.ppat.0030161 |
| [66] |
Zheng Y, Lilo S, Mena P, et al. YopJ-induced caspase-1 activation in Yersinia-infected macrophages:independent of apoptosis, linked to necrosis, dispensable for innate host defense[J]. PLoS One, 2012, 7(4): e36019. DOI:10.1371/journal.pone.0036019 |
| [67] |
Philip NH, Dillon CP, Snyder AG, et al. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling[J]. Proc Natl Acad Sci USA, 2014, 111(20): 7385-7390. DOI:10.1073/pnas.1403252111 |
| [68] |
Ratner D, Orning MP, Starheim KK, et al. Manipulation of interleukin-1β and interleukin-18 production by Yersinia pestis effectors YopJ and YopM and redundant impact on virulence[J]. J Biol Chem, 2016, 291(19): 9894-9905. DOI:10.1074/jbc.M115.697698 |
| [69] |
Schoberle TJ, Chung LK, McPhee JB, et al. Uncovering an Important role for yopJ in the inhibition of caspase-1 in activated macrophages and promoting Yersinia pseudotuberculosis virulence[J]. Infect Immun, 2016, 84(4): 1062-1072. DOI:10.1128/IAI.00843-15 |
| [70] |
Rosadini CV, Zanoni I, Odendall C, et al. A Single bacterial immune evasion strategy dismantles both MyD88 and TRIF signaling pathways downstream of TLR4[J]. Cell Host Microbe, 2015, 18(6): 682-693. DOI:10.1016/j.chom.2015.11.006 |
| [71] |
Ratner D, Orning MP, Proulx MK, et al. The Yersinia pestis effector YopM Inhibits pyrin inflammasome activation[J]. PLoS Pathog, 2016, 12(12): e1006035. DOI:10.1371/journal.ppat.1006035 |
| [72] |
Palace SG, Proulx MK, Szabady RL, et al. Gain-of-function analysis reveals important virulence roles for the Yersinia pestis type Ⅲ secretion system effectors YopJ, YopT, and YpkA[J]. Infect Immun, 2018, 86(9). pii: e00318. https://www.researchgate.net/publication/325714933_Gain_of_Function_Analysis_Reveals_Important_Virulence_Roles_for_the_Yersinia_pestis_Type_III_Secretion_System_Effectors_YopJ_YopT_and_YpkA
|
| [73] |
Chung LK, et al. IQGAP1 is important for activation of caspase-1 in macrophages and is targeted by Yersinia pestis type Ⅲ effector YopM[J]. Mbio, 2014, 5(4): e01402-e01414. |
| [74] |
LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing[J]. Cell Host Microbe, 2012, 12(6): 799-805. DOI:10.1016/j.chom.2012.10.020 |
| [75] |
Chung LK, Park YH, Zheng Y, et al. The Yersinia virulence factor YopM hijacks host kinases to inhibit type Ⅲ effector-triggered activation of the pyrin inflammasome[J]. Cell Host Microbe, 2016, 20(3): 296-306. DOI:10.1016/j.chom.2016.07.018 |
| [76] |
Park YH, Wood G, Kastner DL, et al. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS[J]. Nat Immunol, 2016, 17(8): 914-921. DOI:10.1038/ni.3457 |
| [77] |
Brodsky IE, et al. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type Ⅲ secretion system[J]. Cell Host Microbe, 2010, 7(5): 376-387. DOI:10.1016/j.chom.2010.04.009 |
| [78] |
Zwack EE, Snyder AG, Wynosky-Dolfi MA, et al. Inflammasome activation in response to the Yersinia type Ⅲ secretion system requires hyperinjection of translocon proteins YopB and YopD[J]. Mbio, 2015, 6(1): e02095-e02014. |
| [79] |
Thinwa J, Segovia JA, Bose S, et al. Integrin-mediated first signal for inflammasome activation in intestinal epithelial cells[J]. J Immunol, 2014, 193(3): 1373-1382. DOI:10.4049/jimmunol.1400145 |
| [80] |
Müller AJ, Hoffmann C, Galle M, et al. The S. Typhimurium effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation[J]. Cell Host Microbe, 2009, 6(2): 125-136. DOI:10.1016/j.chom.2009.07.007 |
| [81] |
Suzuki S, et al. Shigella IpaH7. 8 E3 ubiquitin ligase targets glomu-lin and activates inflammasomes to demolish macrophage[J]. Proc Natl Acad Sci USA, 2014, 111(40): E4254-E4263. DOI:10.1073/pnas.1324021111 |
| [82] |
Hermansson AK, Paciello I, Bernardini ML. The orchestra and its maestro:Shigella's fine-tuning of the inflammasome platforms[J]. Curr Top Microbiol Immunol, 2016, 397: 91-115. |
| [83] |
Jeon J, Kim YJ, Shin H, et al. T3SS effector ExoY reduces inflammasome-related responses by suppressing bacterial motility and delaying activation of NF-κB and caspase-1[J]. FEBS J, 2017, 284(20): 3392-3403. DOI:10.1111/febs.2017.284.issue-20 |
| [84] |
Duncan MC, Linington RG, Auerbuch V. Chemical inhibitors of the type three secretion system:disarming bacterial pathogens[J]. Antimicrob Agents Chemother, 2012, 56(11): 5433-5441. DOI:10.1128/AAC.00975-12 |
| [85] |
Lara-Tejero M, Galán JE. Salmonella enterica serovar typhimurium pathogenicity island 1-encodedtype Ⅲ secretion system translocases mediate intimate attachment tononphagocytic cells[J]. Infect Immun, 2009, 77(7): 2635-2642. DOI:10.1128/IAI.00077-09 |
| [86] |
Marshall NC, Finlay BB. Targeting the type Ⅲ secretion system to treat bacterial infections[J]. Expert Opin Ther Targets, 2014, 18(2): 137-152. DOI:10.1517/14728222.2014.855199 |
| [87] |
Gu L, Zhou S, Zhu L, et al. Small-molecule inhibitors of the type Ⅲ secretion system[J]. Molecules, 2015, 20(9): 17659-17674. DOI:10.3390/molecules200917659 |
| [88] |
Anantharajah A, Buyck JM, Sundin C, et al. Salicylidene acylhydrazides and hydroxyquinolines act as inhibitors of type three secretion systems in Pseudomonas aeruginosa by distinct mechanisms[J]. Antimicrob Agents Chemother, 2017, 61(6). pii: e02566-e02616. https://www.ncbi.nlm.nih.gov/pubmed/28396545
|
| [89] |
Guo Z, Li X, Li J, et al. Licoflavonol is an inhibitor of the type three secretion system of Salmonella enterica serovar Typhimurium[J]. Biochem Biophys Res Commun, 2016, 477(4): 998-1004. DOI:10.1016/j.bbrc.2016.07.018 |