




泛素化是真核生物体内重要的蛋白翻译后修饰机制,泛素经泛素活化酶E1活化后,在泛素结合酶E2和泛素连接酶E3的级联作用下传递至底物蛋白,决定底物蛋白的命运[1]。之前人们认为真核生物体内仅存在一种泛素活化酶Uba1(又称Ube1)[2-3],2007年有三篇文章接连报道发现了第二个泛素活化酶Uba6[4-6],两种泛素活化酶E1在将泛素传递至各种不同的泛素结合酶E2时虽有所重叠,但也各具有不同的活性[4]。真核生物体内存在近40个E2和600多个E3[7-8],每个E2可与多个E3反应每个E3可识别多个底物蛋白进行泛素化修饰;多个E2也可与同一个E3反应,不同的E2-E3配对可将不同长度和不同拓扑结构的泛素链传递至底物蛋白上,形成错综复杂的泛素传递网进而修饰胞内蛋白[9]。
鉴于泛素化过程中E1-E2-E3的交叉反应性,目前很难针对某一条特异性的泛素化通路,对其E2-E3间及E3-底物蛋白间的相互作用进行研究,且对于后发现的泛素活化酶Uba6的功能研究仍不够深入。为探究Uba6泛素活化酶介导的泛素化通路,我们已通过正交泛素转移的方法,对比探索了泛素活化酶Uba1与Uba6在哺乳动物细胞中分别对应的泛素化过程,找到了494个在Uba1介导的泛素化通路中对应的底物蛋白,528个在Uba6介导的泛素化通路中对应的底物蛋白及225个二者的共同底物[10]。尽管Uba6与Uba1有着共同作用的泛素结合酶E2,但泛素结合酶USE1被证明是Uba6的特异性E2,与Uba1无交叉反应活性[3],且近期有研究表明,USE1在肺癌细胞中存在过表达现象,可能与肺癌细胞的形成、转移、扩散相关,可作为肺癌发生的生物标记[11]。为进一步探究Uba6-USE1这条特异性泛素化通路的功能,我们在前期研究的基础上,通过RNA干扰技术构建靶向USE1基因的慢病毒载体,包装感染HEK-293细胞,抑制细胞中USE1基因的表达,后期将与过表达USE1基因、正常表达USE1基因细胞株对比其细胞中Uba6-USE1泛素化过程,利用LC-MS液质联用色谱及生物信息学方法找出3条USE1基因表达量不同的泛素化通路中的下游底物,旨为进一步研究Uba6-USE1特异性泛素化通路功能提供实验依据,奠定实验基础。
1 材料与方法 1.1 材料人胚肾HEK-293细胞购自中国科学院细胞库,慢病毒抑制表达载体pLL3.7由上海交通大学药学院孙磊惠赠;Gel/PCR纯化试剂盒、质粒DNA小量抽提试剂盒购自Favorgen公司;限制性核酸内切酶BamH I,Xho I及其缓冲液、T4 DNA连接酶及其缓冲液、anti-USE1抗体购自Thermo Fisher公司;anti-GAPDH抗体购自上海优宁维生物科技股份有限公司;RNA反转录试剂盒购自东洋纺(上海)生物科技公司;大肠杆菌DH5α菌种购自天根生化科技(北京)有限公司;引物的合成及质粒的测序由生工生物工程(上海)股份有限公司完成。
1.2 方法 1.2.1 shRNA的设计与合成根据NCBI数据库中USE1基因的mRNA序列(NM_023079),利用Invitrogen公司提供的BLOCK-iTTM RNAi Designer工具,设计靶向USE1基因的shRNA序列。选择5'端以G开头,GC值在35%-55%之间,且靶点序列起始位置不同的三条序列作为候选,如表 1所示。根据慢病毒抑制表达载体pLL3.7的特点及要求,在寡核苷酸链最两端加上BamH I和Xho I酶切位点,并在5'端加上重建U6启动子的T碱基,在shRNA序列后加上Loop环序列TTCAAGAGA,在3'端加上终止序列TTTTTT。

将合成好的寡核苷酸单链经退火后形成双链,酶切保护碱基后与pLL3.7空载体经T4 DNA连接酶作用于16℃连接过夜,连接产物转化至DH5α感受态细胞后,均匀涂布于含100 μg/mL氨苄青霉素的LB固体平板上,倒置于37℃恒温培养箱过夜,次日挑取单克隆至含相同浓度氨苄青霉素的LB液体培养基中震荡培养过夜,提取质粒,然后进行测序。
1.2.3 细胞的培养冻存的HEK-293细胞经37℃水浴复苏后,加入含10%胎牛血清的DMEM培养基中,在37℃、含5% CO2的培养箱中培养,定期观察细胞状态,根据细胞生长情况适时更换培养基,一般2 d传代一次。
1.2.4 慢病毒质粒共转染HEK-293细胞及慢病毒的包装当HEK-293细胞融合度达70%-80%时准备转染。将3种含不同干扰序列的pLL3.7-shRNA慢病毒重组质粒及作为阴性对照的pLL3.7慢病毒空载体分别与慢病毒包装载体psPAX2、VSVG按照质量比1:3:4混合均匀,加入Opti-MEM培养基;另取相同体积的Opti-MEM培养基与转染试剂PEI进行混合,室温静置5 min后,将分别含有质粒和转染试剂的两管Opti-MEM培养基混匀,室温静置20 min,更换细胞培养基为无血清的DMEM,将混合液加至细胞培养皿中,于37℃、含5%CO2的培养箱中培养6-8sh,更换培养基为完全培养基,继续培养48 h,收集病毒上清液,离心浓缩,冻存于-80℃。
1.2.5 慢病毒滴度的测定及最佳稀释倍数的确定将HEK-293细胞按照每孔2×104个的密度铺于96孔板,培养24 h后,用DMEM完全培养基将浓缩后的病毒上清按10倍递减法依次稀释,培养48 h后,于荧光显微镜下观察病毒感染细胞状况,确定最佳稀释倍数,并按照以下公式计算病毒滴度。
病毒滴度(TU/mL)=荧光细胞数/相应稀释倍数
1.2.6 实时定量PCR及Western Blot检测基因表达量病毒感染前一天,按照每孔5×104个的密度将HEK-293细胞铺于24孔板,次日按照最佳稀释倍数加入病毒稀释液感染细胞,培养24 h后,弃病毒液,更换为DMEM完全培养基,继续培养48 h,收集细胞提取总RNA,反转录为cDNA,以GAPDH为内参,通过实时定量PCR测定目的基因的表达量,其引物序列如下:
USE1-F:5'-ACAGGCCCATTTGACACTCC-3'
USE1-R:5'-CCCATTGCGGTAGAAGTTGG-3'
GAPDH-F:5'-ATCACTGCCACCCAGAAGACT-3'
GAPDH-R:5'-CATGCCAGTGAGCTTCCCGTT-3'
按照上述同样的方法将HEK-293细胞铺于24孔板并进行病毒感染并培养48 h后,每孔加入适量5×蛋白上样缓冲液裂解细胞,收集裂解液并金属浴加热10 min使蛋白变性后进行SDS-PAGE电泳(分离胶浓度为10%),将蛋白条带转印至NC膜后,用5%的脱脂牛奶室温封闭1 h,洗去膜上残留的牛奶,分别用anti-USE1抗体(1:1 000稀释)和anti-GAPDH抗体(1:1 000稀释)敷育,4℃过夜,次日用TNET缓冲液洗膜3次,洗去残留在膜上的一抗,用相应的兔源二抗(1:10 000稀释)室温敷育1 h,用TNET缓冲液洗膜3次后用Odyssey双色红外荧光成像系统扫膜观察结果。
2 结果 2.1 三种USE1抑制表达慢病毒重组质粒pLL3.7-shRNA的鉴定将构建好的质粒送去测序,结果表明插入序列与shRNA设计的目的基因片段完全一致,说明质粒构建成功,可以用于后续实验。
2.2 USE1抑制表达慢病毒重组质粒包装结果USE1抑制表达慢病毒重组质粒pLL3.7-shRNA及包装质粒共转染至HEK-293细胞48 h后,于荧光显微镜(400×)下观察,包装结果如图 1所示。由于pLL3.7抑制表达载体可同时表达GFP绿色荧光蛋白,慢病毒包装后的细胞在蓝色荧光激发下在视野中呈现绿色,其中分图AB、CD、EF和GH分别代表 3种抑制表达重组质粒pLL3.7-shRNA1,2,3及阴性对照pLL3.7慢病毒空载体在同一视野下白光和荧光的包装结果。结果表明,转染48 h后,80%以上的HEK-293细胞均呈现较强绿色荧光,说明3种慢病毒质粒及pLL3.7慢病毒空载体均成功转染至细胞中,即慢病毒包装成功。

|
| 图 1 慢病毒重组质粒转染HEK-293细胞48 h后病毒包装结果(400×) A:pLL3.7-shRNA1白光;B:pLL3.7-shRNA1绿色荧光;C:pLL3.7-shRNA2白光;D:pLL3.7-shRNA2绿色荧光;E:pLL3.7-shRNA3白光;F:pLL3.7-shRNA3绿色荧光;G:pLL3.7 vector白光;H:pLL3.7 vector绿色荧光 |
将收集的慢病毒上清经离心浓缩后,按照10-106倍梯度稀释后感染细胞48 h,在400倍放大的荧光显微镜下观察呈现绿色荧光的细胞个数,计算得三种抑制表达USE1的慢病毒质粒pLL3.7-shRNA1、pLL3.7-shRNA2、pLL3.7-shRNA3的病毒滴度分别为0.7×106 TU/mL,1.0×106 TU/mL和1.5×106 TU/mL,符合滴度要求。同时发现,经10倍、100倍、1000倍稀释的病毒感染细胞后,视野中呈现绿色荧光的细胞数逐渐减少,10倍稀释的病毒感染细胞后仍有约70%的细胞呈现绿色荧光,图 2中A-I分别表示3种pLL3.7-shRNA1,2,3慢病毒包装上清液在稀释10倍、100倍、1 000倍后感染HEK-293细胞在荧光显微镜下的结果。由于浓缩后的病毒上清直接感染细胞48 h后会导致细胞死亡,故选择10倍稀释的病毒上清感染细胞进行后续实验。

|
| 图 2 三种梯度稀释的慢病毒包装液感染HEK-293细胞48 h后结果(400×)s A,B,C:10倍,100倍,1 000倍稀释的pLL3.7-shRNA1慢病毒包装上清液感染细胞,绿色荧光;D,E,F:10倍,100倍,1000倍稀释的pLL3.7-shRNA2慢病毒包装上清液感染细胞,绿色荧光;G,H,I:10倍、100倍、1 000倍稀释的pLL3.7-shRNA3慢病毒包装上清液感染细胞,绿色荧光 |
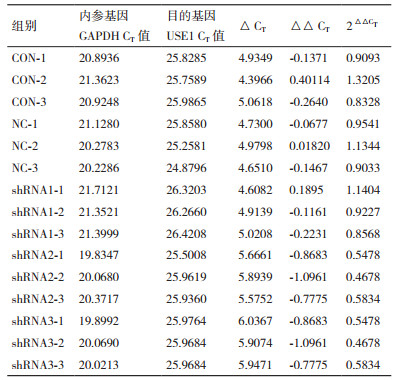
表 2显示了USE1基因的表达量,将表 2中数据经Graph Pad Prism软件处理后得到如图 3所示的结果。从图中可以看出,与未经病毒感染的空白对照CON组相比,包装了pLL3.7-shRNA1质粒的慢病毒上清液感染HEK-293细胞后,USE1基因表达量没有明显减少,差异统计学意义不大(*P > 0.05),而包装了pLL3.7-shRNA2和pLL3.7-shRNA3重组质粒的慢病毒上清感染HEK-293细胞后对USE1目的基因有50%的抑制作用(*P < 0.05),差异具有统计学意义,说明在设计的3条shRNA干扰序列中,shRNA2和shRNA3能够有效抑制HEK-293细胞中USE1基因的表达。

|
| 图 3 RT-PCR检测USE mRNA在HEK-293细胞中的表达(*P < 0.05) |
从感染了10倍稀释慢病毒上清液的HEK-293细胞中裂解蛋白,经Western Blot实验从蛋白水平上检测USE1基因的抑制表达效果,如图 4-A所示。其中1号孔为未经病毒感染的HEK-293细胞中USE1基因正常表达量,2号孔为阴性对照即转染了pLL3.7-vector的HEK-293细胞中USE1基因的表达量,3-5孔分别为感染了包装pLL3.7-shRNA1、pLL3.7-shRNA2、pLL3.7-shRNA3质粒的慢病毒上清后细胞中USE1基因表达水平,从结果中可以看出与1号孔正常HEK-293细胞USE1基因的正常表达量和2号孔阴性对照组相比,3号孔中USE1基因表达量没有明显减少,而4、5号孔中USE1基因表达量明显降低。经灰度量化分析后得到如图 4-B所示的结果,结果进一步表明,相对于空白对照组,包装了pLL3.7-shRNA2和pLL3.7-shRNA3重组质粒的慢病毒上清感染细胞后具有约50%抑制USE1基因表达的效果(*P < 0.01)。

|
| 图 4 Western Blot检测USE1基因在HKE-293细胞中的表达(*P < 0.01) A:Western Blot of USE1 and GAPDH:1:CON; 2:pLL3.7-NC; 3:pLL3.7-shRNA1;4:pLL3.7-shRNA2;5:pLL3.7-shRNA3。B:Protein level of USE1 expression |
泛素化作为普遍存在于真核生物体内的蛋白翻译后修饰机制,在介导内源性蛋白降解、调控细胞周期与细胞凋亡、参与多种信号转导、参与DNA损伤修复及转录调控、协同自噬作用等多个重要的胞内反应中发挥重要作用[12]。泛素化作用底物繁多且通路网络复杂,当其机制出现异常,往往会导致胞内蛋白水平失衡,胞内多种生理活动不能正常进行,进而引发包括肿瘤、神经退行性疾病、炎症及风湿性疾病等[13-15],因此,维持泛素化正常进行对于更新胞内蛋白、清除衰老及错误折叠蛋白、维持机体内环境稳定具有重大意义[16]。由于泛素结合酶E2和泛素连接酶E3成员众多,彼此间作用相互交错导致目前对某条特异的泛素化通路中,E2-E3及E3-底物蛋白间的具体的相互作用仍然模糊。而第二个泛素活化酶Uba6自发现以来,对其功能的认识依然不足,而对其特异性泛素结合酶USE1的研究更少,对这条特异性泛素化通路的研究对发现泛素活化酶Uba6功能、发现潜在有价值的底物蛋白、丰富泛素化理论等方面具有重要意义。
慢病毒载体是以人类免疫缺陷I型病毒HIV-1为基础建立的,对分裂细胞和非分裂细胞均有强感染力的一种载体,被广泛应用于RNA干扰技术、基因治疗等研究中[17-18]。当慢病毒进入细胞后,病毒RNA反转录进入细胞核中,可稳定整合至宿主基因组上,通过转染至细胞中可产生表达shRNA的高滴度病毒,实现在多种细胞中特异、稳定的基因沉默效果[19-20]。根据靶标基因的序列设计的shRNA可通过克隆到慢病毒表达载体上,经转录后折叠形成发卡结构,在细胞内进一步加工成siRNA,诱导同源靶基因的mRNA降解,发挥RNA干扰作用,沉默靶标基因的表达[21]。
基于以上理论基础,本实验在前期探索泛素活化酶Uba1和Uba6分别对应的特异性泛素化通路研究的基础上,加入了对Uba6的特异性泛素结合酶USE1研究,利用慢病毒载体干扰技术,设计3条靶向USE1基因的shRNA干扰序列,构建3种抑制USE1基因表达的慢病毒载体,经慢病毒包装、感染细胞后检测其抑制USE1基因表达情况,得到能够有效抑制USE1基因表达的细胞株。本实验成功构建了3种序列正确的USE1基因抑制表达慢病毒载体,经mRNA水平及蛋白水平检测,其中2种慢病毒重组质粒经包装转染至HEK-293细胞后,可有效抑制USE1基因的表达,编号分别为shRNA-2、shRNA-3,病毒包装液感染细胞后可得抑制USE1基因表达的细胞株,为后续实验中对比、寻找过表达USE1基因和正常表达USE1基因细胞株中泛素化对应的下游底物奠定基础,也为进一步研究Uba6-USE1这条特异性泛素化通路的功能提供依据。
4 结论本研究基于泛素活化酶Uba6及其特异性泛素结合酶USE1的泛素化通路,为得到USE1基因的抑制表达细胞系,设计了3条靶向USE1基因的shRNA序列,成功构建了3种RNA干扰USE1基因表达的慢病毒重组质粒,经慢病毒包装感染HEK 293细胞后检测蛋白表达情况,最终确定并得到了其中2种抑制USE1基因表达率在50%左右的shRNA序列及其慢病毒包装上清液,病毒上清感染细胞后即可得到抑制USE1基因表达的细胞株。
| [1] |
Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases[J]. Nature Reviews Molecular Cell Biology, 2009, 10(6): 398-409. DOI:10.1038/nrm2690 |
| [2] |
Pickart CM, Eddins MJ. Ubiquitin:structures, functions, mechanisms[J]. Biochimica Biophysica Acta.Molecular Cell Research, 2004, 1695(3): 55-72. |
| [3] |
Pickart CM. Back to the future with ubiquitin[J]. Cell, 2004, 116(2): 181-190. DOI:10.1016/S0092-8674(03)01074-2 |
| [4] |
Jin J, Li X, Gygi SP, et al. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging[J]. Nature, 2007, 447(7148): 1135-1138. DOI:10.1038/nature05902 |
| [5] |
Chiu YH, Sun Q, Chen ZJ. E1-L2 activates both ubiquitin and FAT10[J]. Mol Cell, 2007, 27: 1014-1023. DOI:10.1016/j.molcel.2007.08.020 |
| [6] |
Pelzer C, Kassner I, Matentzoglu K., et al. UBE1L2, a novel E1 enzyme specific for ubiquitin[J]. Journal of Biological Chemistry, 2007, 282(32): 23010-23014. DOI:10.1074/jbc.C700111200 |
| [7] |
Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance[J]. Journal of Cell Science, 2012, 125(Pt 3): 531. |
| [8] |
Zhou MJ, Chen FZ, Chen HC. Ubiquitination involved enzymes and cancer[J]. Medical Oncology, 2014, 31(8): 93. DOI:10.1007/s12032-014-0093-6 |
| [9] |
Zhao B, Bhuripanyo K, Zhang KY, et al. Orthogonal ubiquitin trans-fer through engineered E1-E2 cascades for protein ubiquitination[J]. Chemistry & Biology, 2012, 19(10): 1265-1277. |
| [10] |
Liu X, Zhao B, et al. Orthogonal ubiquitin transfer identifies ubiqu-itination substrates under differential control by the two ubiquitin activating enzymes[J]. Nat Commun, 2017, 8: 14286. DOI:10.1038/ncomms14286 |
| [11] |
Kim SJ, Lee TH, Sang HN, et al. Association of Uba6-Specific-E2(USE1) With Lung Tumorigenesis[J]. Journal of the National Cancer Institute, 2016, 109(3): 1-11. |
| [12] |
Hershko A, Ciechanover A. The ubiquitin system[J]. Annual Review of Biochemistry, 1998, 67: 425-479. DOI:10.1146/annurev.biochem.67.1.425 |
| [13] |
王佳悦, 赵博. 泛素化在肿瘤治疗方面的应用及展望[J]. 生命的化学, 2017, 37(6): 879-887. |
| [14] |
何伟珍, 徐晓东. 泛素-蛋白酶体途径与疾病[J]. 海南医学, 2007, 18(3): 141-142. DOI:10.3969/j.issn.1003-6350.2007.03.082 |
| [15] |
唐珍, 罗蓉. 泛素-蛋白酶体通路与疾病发生的研究进展[J]. 兰州大学学报:医学版, 2014, 40(3): 71-75. |
| [16] |
Dou QP, Zonder JA. Overview of proteasome inhibitor-based anti-cancer therapies:perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system[J]. Current Cancer Drug Targets, 2014, 14(6): 517-536. DOI:10.2174/1568009614666140804154511 |
| [17] |
Sumimoto H, Kawakami Y. Lentiviral vector-mediated RNAi and its use for cancer research[J]. Future Oncol, 2007, 3(6): 655-664. DOI:10.2217/14796694.3.6.655 |
| [18] |
Katayama K, Koichiro W, Miyoshi H, et al. RNA interfering approach for clarifying the PPAR pathway using lentiviral vectors expressing short hairpin RNA[J]. FEBS Letters, 2004, 560(3): 178-182. |
| [19] |
Kafri T, Praag HV, Ouyang L, et al. A Packaging cell line for lentivirus vectors[J]. Journal of Virology, 1999, 73(1): 576. |
| [20] |
Naldini L, Blömer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector[J]. Science, 1996, 272(5259): 263-267. DOI:10.1126/science.272.5259.263 |
| [21] |
张景海, 杨保胜, 颜真, 等. 药学分子生物学[M]. 第4版. 北京: 人民卫生出版社, 2013.
|