




地黄是我国常用中药材之一,它是玄参科地黄属多年生草本植物,地黄根作为药材,具有显著的药用效果,是多种中成药的成分之一,具有很高的经济价值[1]。但是,市场上流通的地黄来源杂乱,品质不一,且不同品种间药用成分差异较大[2]。所以,应采取有效的方法鉴定优良种质的地黄品种。
DNA分子标记技术是基因定位克隆,遗传图谱构建等研究工作的重要工具,目前已广泛应用于作物分子辅助育种及种质资源多样性分析等领域[3-4],特别是单核苷酸多态性研究备受关注。单核苷酸SNP是单个核苷酸变异而产生的DNA水平上的多态性。SNP是大多数基因组中最丰富和稳定的遗传变异形式,多态性高,数量多,已成为植物分子辅助育种的主要分子标记之一[5]。
基于转录组测序能够大规模的挖掘SNP位点。在植物中的应用也十分广泛,候莉娟等[6]在实验室已有转录组数据的基础上,挖掘了大量羊草的SNP对羊草种质进行分型;周军永等[7]以李府贡枣不同处理枣果实的转录组序列为基础,分析了转录组数据中SNP位点的分布,为枣的遗传结构和遗传分化奠定了基础。Wu等[8]基于银杏转录组测序收集并验证SNP位点,并分析群体遗传多样性,为银杏的遗传和基因组研究提供了有用资源。
目前,利用SNP分子标记技术探讨地黄种属亲缘关系及品种鉴定的研究很少,本研究利用RNA-seq技术,对85-5地黄新鲜块根进行转录组测序,根据测序数据与NCBI中SRA数据库中的3个地黄数据比对,利用Samtools(http://samtools.sourceforge.net/)和VarScan v.2.2.7(http://varscan.sourceforge.net/)软件寻找候选SNP。针对候选SNP位点设计引物,选择能够扩增出单一目的条带的引物,对地黄28个品种间扩增,旨为地黄遗传多样性分析和品种鉴定奠定基础。
1 材料与方法 1.1 材料实验用地黄种质收集于河南4个市县、山东2个区县和上海华东师范大学,包括裂叶地黄(Rehmannia piasezkii)和地黄(Rehmannia glutinosa)这2个种,共28份种质。均经过ITS测序与NCBI比对,确定为裂叶地黄或地黄种质[9](表 1)。

实验用Taq Master Mix由北京康为世纪生物科技有限公司合成,具有稳定性好、灵敏度高、快速简便、特异性强等优点。
SNP引物Primer软件设计后于英潍捷基(上海)贸易有限公司合成。
1.2 方法 1.2.1 挖掘SNP位点根据实验室前期得到的地黄转录组数据,将其与在NCBI数据库上下载的3个转录组数据库,下载的转录组数据测序数据分别是由河南农业大学上传的SRR444423.sra数据量为485 M;河南农业大学上传SRR832972.sra数据量2.1 G;药用植物研究所上传的SRR832973.sra数据量2.1 G。然后通过格式转换之后,以这3个组装好的转录本为模板序列,将原始序列与其进行比对,利用Samtools(http://samtools.sourceforge.net/)和VarScan v.2.2.7(http://varscan.sourceforge.net/)软件寻找候选SNP。
1.2.2 设计SNP引物根据挖掘到的候选SNP位点,利用Primer软件设计引物。在SNP位点的上下游分别设计正向引物和反向引物,理想引物的长度为18-24 bp,Tm值为50-60℃,能保证上下游引物的Tm值一般不超过2℃;引物序列的GC含量一般为40%-60%,过高或过低都不利于引发反应,上下游引物的GC含量不能相差太大。
1.2.3 筛选SNP引物通过PCR来验证SNP引物。设计的SNP引物,需要确保能扩增出单一、明亮的条带。PCR扩增反应体系为20 μL,包含6 μL的dd H2O,1 μL的模板DNA,1 μL上游引物,1 μL下游引物,10 μL Mix。扩增程序:94℃预变性3 min;94℃变性30s,适宜退火温度退火30s,72℃延伸30 s,34个循环;72℃延伸5 min;4℃保存。采用1%琼脂糖凝胶电泳检测PCR扩增产物,胶回收后测序。
1.2.4 SNP位点用于28种地黄分型分别以28种地黄基因组DNA为模板,选用初筛的引物来对其进行PCR扩增,回收目的片段,Sanger法正反向测序目的序列,检测SNP位点多态性。根据SNP位点绘制指纹图谱。
2 结果 2.1 地黄SNP位点的挖掘在地黄转录组中共有102 075条Unigene中,检测到35 339个SNP位点,在转录组数据中的发生频率为0.51/kb。检测出的SNP位点中发生转换共22 718个(64.28%),颠换共12 621个(35.72%),这6种单核苷酸变异里C/T和A/G的频率最高,分别达到31.95%和32.33%,其他4种单核苷酸变异中A/T、A/C、T/G、C/G的频率分别为9.22%、9.36%、8.80%和8.34%。本次研究极大丰富了地黄的SNP位点信息(表 2)。

根据SNP挖掘结果,用Primer 5设计了39对引物,包含了54个候选SNP位点。对设计的39对SNP引物,以地黄85-5基因组为模板进行PCR扩增,经琼脂糖凝胶电泳检测,从中挑选出了28对符合预期产物长度、扩增效果清晰单一的SNP引物对(图 1),用以进行后续分型实验。

|
| M:分子量标准;1-20:表示不同引物扩增出的条带 图 1 引物特异性筛选部分结果 |
为开发多态性地黄SNP引物,我们用2.2实验筛选出的28对特异性引物对28种地黄种质进行分型实验(图 2)。经过PCR扩增目的片段,切胶纯化回收后用Sanger法直接测序。在28对引物扩增的PCR产物中,能够直接测序成功并且SNP位点在不同地黄种质间不同的仅有7对(表 3)。经过DNAMAN序列比对软件比对后,可以直观的看出不同地黄种质间的SNP位点变化,位点SNP1的比对图如图 2所示。经测序后统计7对引物所包含的8个SNP位点在28种地黄种质间的检测结果,如表 4所示。


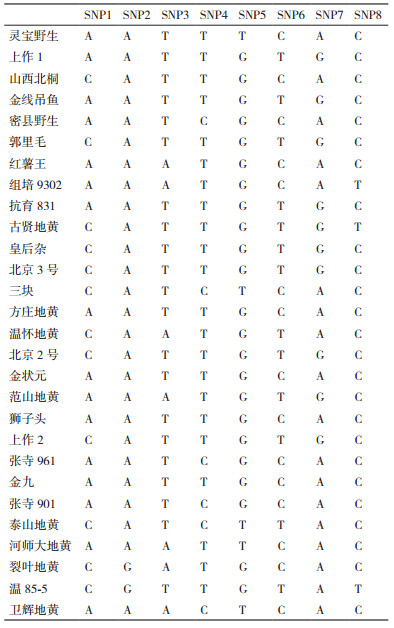
通过对28份地黄的8个SNP位点的分析,发现可以将地黄17个种质区分开。利用这8个SNP位点绘制17份地黄种质的指纹图谱,如图 3所示,不同引物对的分型结果用不同的颜色区分;同一引物对的分型结果用数据条长短区分,数据条长度相同的种质,基因型一致,数据条长度不同的种质,表明在基因分型时被分到不同的组。如DH2和DH3两份地黄种质仅在SNP3位点存在差异,其余的SNP位点均无差异,这表明两份种质的差异较小;而DH6和DH17则是除了在SNP7变异位点处相同,其余的6个SNP位点均存在差异,表明它们差异较大。

|
| DH1:上作1、金线吊鱼、抗育831;DH2:山西北桐;DH3:方庄地黄、金状元、狮子头、金九;DH4:范山地黄;DH5:密县野生、张寺961、张寺901;DH6:卫辉地黄;DH7:灵宝野生;DH8:河师大地黄;DH9:组培9302;DH10:山西北桐;DH11:古贤地黄;DH12:皇后杂、北京2号、上作2、郭里茂、北京3号;DH13:三块;DH14:温怀地黄;DH15:泰山地黄;DH16是裂叶地黄;DH17:85-5地黄 图 3 利用8个SNP位点对地黄17个种质绘制的指纹图谱 |
目前,应用于地黄的分子标记主要有RAPD、ISSR[10]、ITS[9]、SSR[11-12]和SCoT[1]等。但这些分子标记各有不足[13],如技术难度高,实验重复性差,对PCR体系要求高等,限制了这些技术的发展与应用。SNP分子标记是一种特异性强的遗传标记,随着高通量测序技术的广泛应用,以及SNP标记的检测成本的逐渐下降,基于转录组测序来挖掘SNP位点,会使得SNP分子标记技术越来越高效。对于多倍体或没有参考基因组的物种来说,利用新一代高通量转录组测序技术可大量鉴定SNP[6]。近年也被成功地应用于植物的基因分型、品种鉴定、新品种选育等研究中,Jiang[14]利用开发的14个SNP标记对30个柑橘品种进行基因分型,区分了品种间的亲缘关系;李志远等[15]在甘蓝中筛选出50个核心SNP标记用于构建59个甘蓝品种的指纹图谱,能有效鉴定品种的特异性和真实性;Distefano等[16]开发出了21个柑橘SNP标记,并对18个柑橘品种进行遗传多样性分析;吴澎等[17]在小麦中可利用SNP标记技术和基因工程技术相结合,将多个优质感官性状基因聚合到一个小麦新品种上;Ferguson等[18]利用53个木薯品种对候选SNP位点的验证,筛选出1 190个稳定且具有多态性的SNP标记,用于对木薯的多样性评估和地理起源分析。
目前,常用于SNP分析的有等位基因特异性PCR法[19]、高分辨率溶解曲线分析[20]、DNA芯片法[21]和直接测序分型法等。由于AS-PCR法对PCR体系要求严格,HRMA法需要使用专门的设备,DNA芯片法需要大量合成特异性探针,都不是最优的SNP分型法。基于Sanger测序法的SNP分型,它的测序准确度高,不仅能够发现和检测不同个体间存在的所有碱基变异,同时还可以明确各个的位置及变异类型,是实验室常用的SNP分型方法。
但是,到目前为止,还未见使用SNP分子标记对地黄种质进行研究。SNP分子标记因其在基因组中数量最多、分布最广及具有双等位基因等特性,正好可以打破地黄品种的遗传基础狭窄,品种间遗传分化不明显[22]这种局限性,SNP标记将在新品种的特异性、真实性鉴定方面有更多应用。为此,本研究利用首次使用SNP分子标记分析28种地黄的遗传多样性,丰富地黄分子标记技术,为区分地黄种质提供有力的分子标记技术。
本研究通过转录组挖掘SNP位点,丰富了地黄基因组SNP变异信息,在Unihene中共发现3 339个SNP位点,其中转换类型发生频率显著高于颠换类型,转换发生频率约为颠换的两倍,这与其他植物转录组SNP变异类型的比例相似[23-24]。转换类型中A/G(32.33%)和C/T(31.95%)的发生频率最高,颠换类型中A/T最高为9.22%。地黄转录组中SNP位点非常丰富,可为地黄遗传多样性分析、亲缘关系鉴定与品种区分等方面提供丰富的基础数据信息。根据SNP候选位点设计的39对引物中,以地黄85-5基因组为模板进行PCR扩增,筛选出28对扩增条带单一、产物长度符合预期的引物,引物特异性筛选率为72%,成功率较高。但是筛选出的候选SNP位点验证成功率并不是很高,仅有7对引物所包含的8个SNP位点在28种地黄种质中有差异。这可能是由于在转录组挖掘SNP位点时,对原始测序数据分析时造成的SNP位点的假阳性的原因[25-26]。使用28对引物在28种地黄中进行分型实验,结果仅有7对引物所包含的8个SNP位点表现出多态性,根据8个SNP位点构建纹图谱,可以直观的区分17种地黄种质。根据指纹图谱可以看出8个SNP位点可以将裂叶地黄从其余27种地黄种质中区分出来,表明筛选出SNP位点可以用于地黄的种间区分。其余的鉴别出来的16种地黄种质都属于地黄种,表明8个SNP位点也可用于地黄的种间区分。
在地黄其他分子标记的研究中,张进忠等[27]筛选出的17条RAPD引物在10个地黄品种都可以扩增出各自的多态性条带,检测10个地黄品种的种质遗传多样性;周延清[10]筛选出17条RAPD引物和10条ISSR引物用于对10个地黄品种的种质遗传多样性分析,并且筛选出能够鉴别10个地黄样品的RAPD引物两个与ISSR引物一个;郭冠瑛[2]利用地黄转录组信息,设计了320对EST-SSR引物,213对引物在地黄85-5的基因组扩增出了产物,只有80对引物在36份地黄种质中具有多态性。杨珂等[1]利用5个SCoT引物构建30份地黄种质的指纹图谱,结果能区分开7个地黄常见栽培品种。我们基于8个SNP位点绘制指纹图谱,可以将28个地黄种质区分出17种,区分力度稍弱,这有待于继续大量开发SNP位点,为地黄品种鉴定提供更有效的技术。
本研究以鉴定地黄品种,丰富地黄分子标记为目的,采用PCR产物直接测序的方法进行地黄的SNP分型,结果表明有7对引物,包含8个SNP位点能在17份地黄材料中表现出多态性,并根据8个SNP位点的差异构建了指纹图谱,对地黄品种的遗传多样性做了补充,为鉴定地黄品种开发了有效的分子标记,为大量开发地黄SNP标记来全面评估地黄遗传多样性奠定基础,使用SNP分子标记技术来准确鉴定地黄品种对种质管理和育种也有重要意义。
4 结论通过地黄转录组挖掘SNP位点,在28份地黄种质中筛序多态性SNP位点,利用8的SNP位点能够鉴定17种不同的地黄种质。
| [1] |
杨珂, 周延清, 段英, 等. 地黄SCoT分子标记体系的建立和指纹图谱的构建[J]. 广西植物, 2019, 39(5): 608-614. |
| [2] |
郭冠瑛.地黄大容量转录组文库的构建及EST-SSR标记的开发与鉴定[D].郑州: 河南农业大学, 2013. http://cdmd.cnki.com.cn/Article/CDMD-10466-1015505673.htm
|
| [3] |
周延清, 王婉珅, 王向楠, 等. 地黄DNA分子标记与基因功能研究进展[J]. 植物学报, 2015, 50(5): 665-672. |
| [4] |
王丰青, 谢彩侠, 孙瑞斌, 等. 地黄种质创新与品种选育研究进展[J]. 中国中药杂志, 2018, 43(21): 4203-4209. |
| [5] |
Laosatit K, Tanya P, Somta P, et al. De novo transcriptome analysis of apical meristem of Jatropha spp[J]. using 454 pyrosequencing platform, and identification of SNP and EST-SSR markers, Plant Molecular Biology Reporter, 2015, 34(4): 786-793. |
| [6] |
侯莉娟, 齐晓, 齐冬梅, 等. 用于羊草基因分型的SNP分子标记技术研究[J]. 草业学报, 2016, 25(2): 105-113. |
| [7] |
周军永, 陆丽娟, 刘茂, 等. 基于李府贡枣转录组测序的SSR和SNP特征分析[J]. 江苏农业科学, 2019, 47(4): 51-54. |
| [8] |
Wu YQ, Zhou Q, Huang SJ, et al. SNP development and diversity analysis for Ginkgo biloba based on transcriptome sequencing[J]. Trees, 2019, 33(2): 587-597. |
| [9] |
王婉珅.地黄DNA条形码序列的筛选与鉴定研究[D].新乡: 河南师范大学, 2016. http://cdmd.cnki.com.cn/Article/CDMD-10476-1016231974.htm
|
| [10] |
周延清, 景建洲, 李振勇, 等. 利用RAPD和ISSR分子标记分析地黄种质遗传多样性[J]. 遗传, 2004(6): 922-928. |
| [11] |
Li M, Yang Y, Feng F, et al. SSR development and utilization with rehmannia glutinosa transcriptome[J]. International Journal of Agriculture & Biology, 2015, 18(1): 55-56. |
| [12] |
Li XJ, Chao J, Ning X, et al. Sorting and identification of, rehmannia glutinosa, germplasm resources based on EST-SSR, scanning electron microscopy micromorphology, and quantitative taxonomy[J]. Industrial Crops and Products, 2018, 123: 303-314. |
| [13] |
周琳, 段玉, 文博, 等. SNP分子标记及其在木本植物遗传育种的应用[J]. 亚热带植物科学, 2018, 47(2): 187-193. |
| [14] |
Jiang D, Ye QL, Wang FS, et al. The mining of citrus est-snp and its application in cultivar discrimination[J]. Agricultural Sciences in China, 2010, 9(2): 179-190. |
| [15] |
李志远, 于海龙, 方智远, 等. 甘蓝SNP标记开发及主要品种的DNA指纹图谱建[J]. 中国农业科学, 2018, 51(14): 2771-2788. |
| [16] |
Distefano G, Malfa SL, Alessandra G, et al. EST-SNP genotyping of citrus species using high-resolution melting curve analysis[J]. Tree Genetics & Genomes, 2013, 9(5): 1271-1281. |
| [17] |
吴澎, 刘娟, 田纪春. 单核苷酸多态性(SNP)分子标记在小麦遗传育种中的研究进展[J]. 农学学报, 2019, 9(1): 54-58. |
| [18] |
Ferguson ME, Hearne SJ, Close TJ, et al. Identification, validation and high-throughput genotyping of transcribed gene SNPs in cassava[J]. Theoretical & Applied Genetics, 2012, 124(4): 685-695. |
| [19] |
Hansson B, Kawabe A. A simple method to score single nucleotide polymorphisms based on allele-specific PCR and primer-induced fragment-length variation[J]. Molecular Ecology Resources, 2010, 5(3): 692-696. |
| [20] |
Liew M. Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons[J]. Clinical Chemistry, 2004, 50(7): 1156-1164. |
| [21] |
Wang D, Parkman HP, Jacobs MR, et al. DNA microarray snp associations with clinical efficacy and side effects of domperidone treatment for gastroparesis[J]. Journal of Biomedical Informatics, 2012, 45(2): 316-322. |
| [22] |
夏至, 黄勇, 李贺敏, 等. 基于核基因ITS及叶绿体psbA-trnH和trnS-trnG基因怀地黄栽培起源探讨[J]. 中草药, 2018, 49(2): 423-430. |
| [23] |
Lu FH, Yoon MY, Cho YI, et al. Transcriptome analysis and SNP/SSR marker information of red pepper variety YCM334 and Taean[J]. Scientia Horticulturae, 2011, 129(1): 38-45. |
| [24] |
化文平, 韩立敏, 魏磊, 等. 基于盾叶薯蓣转录组的SNP和SSR位点分析[J]. 分子植物育种, 2017, 15(10): 4003-4009. |
| [25] |
吴欣.基于转录组测序的石磺科贝类SSR和SNP两种分子标记的开发[D].上海: 上海海洋大学, 2016. http://cdmd.cnki.com.cn/Article/CDMD-10264-1016912398.htm
|
| [26] |
Wang B, Tan HW, Fang W, et al. Developing single nucleotide polymorphism(SNP)markers from transcriptome sequences for identification of longan(Dimocarpus longan)germplasm[J]. Horticulture Research, 2015, 2: 14065. |
| [27] |
张进忠, 王永芬, 王建波. 地黄品种遗传多样性RAPD分析[J]. 河南农业科学, 2006(6): 97-100. |