




2. 河北省科学院生物研究所,石家庄 050081;
3. 河北省主要农作物病害微生物控制工程技术研究中心,石家庄 050081
2. Institute of Biology, Hebei Academy of Science, Shijiazhuang 050081;
3. Main Crops Disease of Microbial Control Engineering Technology Research Center in Hebei Province, Shijiazhuang 050081
环状芽孢杆菌(Bacillus circulans)是一类革兰氏阳性菌,通常从土壤、污水、食物和伤口中分离得到[1]。这种细菌与人类感染有关,包括败血症、混合脓肿感染和伤口感染[2]。此外,研究发现环状芽孢杆菌中能够合成几丁质酶[3]、环糊精糖基转移酶[4-5]、木聚糖酶[6]、半乳糖苷酶[7]等工业用酶,也有研究发现环状芽孢杆菌对铝土矿的分解具有协同作用[8]。McLeod[9]从环状芽孢杆菌中发现并鉴定到环杆菌素(Circulin),Dion等[10]从环状芽孢杆菌NRRL B3312和B3313菌株中发现并鉴定到丁酰苷菌素(Butirosin),这两种物质作为抗生素一直使用至今; He等[11]从环状芽孢杆菌J2154中鉴定出多种环脂肽类抗菌物质(Circulocins α-δ),这些物质对革兰氏阳性菌具有很好的抑制效果; Das等[12]对一株海洋来源的环状芽孢杆菌鉴定进行研究,发现其代谢产物中含有具有抑菌活性的脂肽类生物表面活性剂; 王美琴等[13]发现环状芽孢杆菌Jcxy8对灰霉病菌具有拮抗作用,其发酵上清对灰霉病菌的生长具有显著抑制作用,这些研究说明环状芽孢杆菌具有一定的抗菌物质合成能力。
虽然已有大量的环状芽孢杆菌相关研究,但到目前为止,只有9株环状芽孢杆菌完成了基因组测序。Tettelin等[14]在2005年提出泛基因组(Pangenome)的概念,它包括核心基因组(由所有菌株共有的基因组成)、非必须基因组(由一个或多个菌株缺失的基因组成)和菌株的特殊基因(由每个菌株特有的基因组成)。近年来,泛基因组分析在微生物基因组功能基因鉴定中得到了广泛的应用[15]。与比较基因组学经典分析不同,泛基因组分析可以预测蛋白质编码基因的数量,并在新的环状芽孢杆菌基因组测序时,通过计算可以预测整个环状芽孢杆菌泛基因组的大小[16]。泛基因组分为开放的和封闭的两种类型,开放的泛基因组是指有的物种在有新的菌株测序时核心基因组中会有新的基因出现; 封闭的泛基因组是指有的物种在有新的菌株测序时核心基因组中不会有新的基因出现[17]。对菌株代谢产物进行鉴定时,有时传统的分离纯化鉴定手段受到局限,通过对已知基因组数据进行挖掘,找到一些新型的次级代谢产物合成通路,通过对这些通路的鉴定,有可能发现新型的活性物质[18]。
在本研究中,我们从NCBI找到了目前所有的9个环状芽孢杆菌基因组,并对其进行了泛基因组分析,确定了其泛基因组特征; 并通过预测软件对次级代谢产物合成通路进行了挖掘,确定了其最可能具有的活性物质合成能力。本研究旨在为进一步探索环状芽孢杆菌的基因组信息,为对该菌的进一步利用奠定基础。
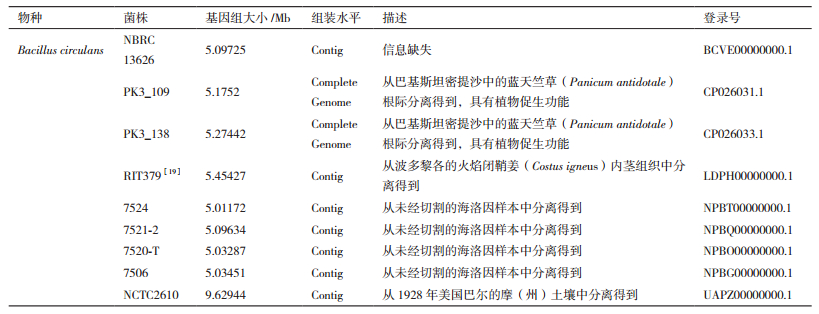
1 材料与方法 1.1 材料在NCBI数据库(https://www.ncbi.nlm.nih.gov/)中的“Genome”条目下输入“Bacillus circulans”查找环状芽胞杆菌属所有菌株的的NCBI登录号。本研究中使用的9株环状芽孢杆菌的基因组均检索自2019年3月之前在GenBank数据库中提交的基因组,详细信息如表 1所示。
下载表 1中菌株对应的gbk格式基因组数据,使用REALPHY(http://realphy.unibas.ch)进行全基因组比对[20],数据以gbk格式进行上传,使用默认参数运行。使用MEGA X[21]构建系统进化树,选择最大似然(ML)算法以及Kimura 2参数模型[22]进行计算,设置重复抽样次数为1 000,其它参数使用默认值。
1.2.2 泛基因组和核心基因组分析下载表 1中菌株对应的基因组组装及注释等全部数据,选择cds_from_genomic.fna.gz,protein.faa.gz和feature_table.txt.gz三种后缀的文件作为数据输入文件,使用PGAweb[23]软件的PGAP模块[24]选择GF(Gene Family,基因簇)算法对9个基因组进行分析,BLAST参数最小值设为50,E-value设为1e-10,全局匹配区不少于蛋白质序列的50%,一致性也不低于50%。
下载结果文件,选择Orthologs_Cluster1.txt文件,使用PanGP[25]进行数据拟合。算法选用遍历所有组合(Traverse all),其他值使用默认值。
1.2.3 次级代谢产物合成基因簇分析利用antiSMASH[26](https://antismash.secondarymetabolites.org/)的细菌分析模块对9株环状芽孢杆菌次级代谢产物生物合成基因簇进行预测注释,参数选用默认值。
2 结果 2.1 环状芽孢杆菌基因组基本数据分析截止至2019年3月20日,NCBI数据库中共有9个环状芽孢杆菌基因组,其中2个组装到完整基因组水平。通过统计发现,9株环状芽孢杆菌的基因组大小在5.01-9.63 Mb之间,GC含量在35.3%-39.8%之间,预测基因数在4 756-9 553个之间,均没有质粒数据报道,数据释放时间均在2015年之后。
2.2 基因组系统发育分析使用REALPHY将9株环状芽孢杆菌基因组进行了比对,然后利用MEGA X选择最大似然法,构建了系统进化树。结果(图 1)显示,9株环状芽孢杆菌在进化树上被归为了两个分支,其中NBRC 13626、NCTC2610、RIT379、7520-T、7524、7506五株菌为一个亚支,7521-2、PK3_109、PK3_138三株菌为一个亚支。与分离源数据比较发现,部分菌株的进化关系与分离源具有一定相关性,从巴基斯坦沙漠分离的两株菌(PK3_109、PK3_138)均在一个亚支,但从海洛因样本中分离的四株菌(7520-T、7524、7506和7521-2)却在两个亚支。

|
| 图 1 九株环状芽孢杆菌的系统进化树 |
环状芽孢杆菌的基因组大小和基因数目成正比,菌株NCTC2610的基因组最大,其对应的基因数也最多。对环状芽孢杆菌进行了泛基因组和核心基因组分析,共有47 354个蛋白质编码基因被分为9 572个基因家族,每个基因家族代表一个假定的同源基因。每个基因家族存在于不同的基因组,一个基因家族覆盖的基因组越多,这个基因家族中的基因就越保守。核心基因组中的基因决定了主要表型性状的基本生物学特性[15]。在9个基因组中,鉴定出3 622个基因家族组成核心基因组,其占环状芽孢杆菌泛基因组的37.83%。鉴定出特有基因4 593个,占泛基因组的47.98%;其中菌株NCTC2610的特有基因最多,为3 031个; 菌株NBRC 13626的特有基因最少,为39个(图 2)。

|
| 图 2 环状芽孢杆菌基因组中基因家族出现的频率 |
在泛基因组分析的基础上,我们使用PanGP软件计算了泛基因组、核心基因组和基因组数目之间的关系(图 3)。泛基因组大小(P)与基因组数(G)关系的拟合方程为P=1364.59G0.68+3491.53(R2=0.999 7),通过拟合方程可以看出环状芽孢杆菌的泛基因组大小随着测序基因组数目的增加而增大。核心基因组大小(C)与基因组数(G)关系的拟合方程为C=3341.67e-1.01G+3621.87(R2=0.983 952),根据该拟合方程可以看出环状芽孢杆菌的核心基因组大约由3 622个基因家族组成。随着基因组测序数量的增加,泛基因组随之增加,而核心基因组则逐渐减少。因此,我们可以推测环状芽孢杆菌的泛基因组是开放的。

|
| 图 3 环状芽孢杆菌泛基因组和核心基因组特征 |
在上述数据的基础上,使用PanGP软件计算了新基因家族数量(N)与基因组数(G)的关系(图 4),并计算出拟合方程为N=1062.09G-0.36(R2=0.984 796)。分析表明环状芽孢杆菌具有开放的泛基因组,在目前基因组数量水平上,当添加新测序的基因组后,可以计算出其泛基因组的基因家族数量预计增加451个,核心基因组的基因家族数量预计是3 622个,新基因家族数量预计会增加463个。

|
| 图 4 环状芽孢杆菌新基因数量与基因组的关系 |
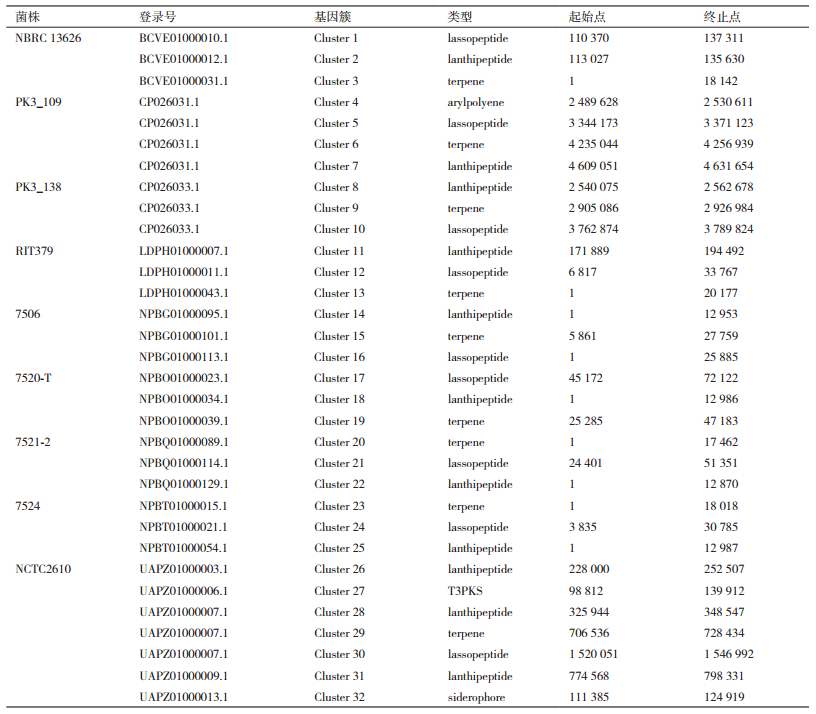
利用在线预测软件antiSMASH对9株环状芽孢杆菌基因组中的次级代谢产物合成基因簇进行预测,结果共注释到6类、32个次级代谢基因簇(表 2)。其中,注释到的基因簇有1个芳基多烯(Arylpolyene)、1个含铁细胞(Siderophore)、1个第3类聚酮类化合物(T3pks)、9个套索肽(Lassopeptide)、9个萜烯(Terpene)和11个羊毛硫肽(Lantipeptide)基因簇。这说明,目前测序的环状芽孢杆菌的主要代谢产物可能是羊毛硫肽、套索肽和萜烯类化合物。
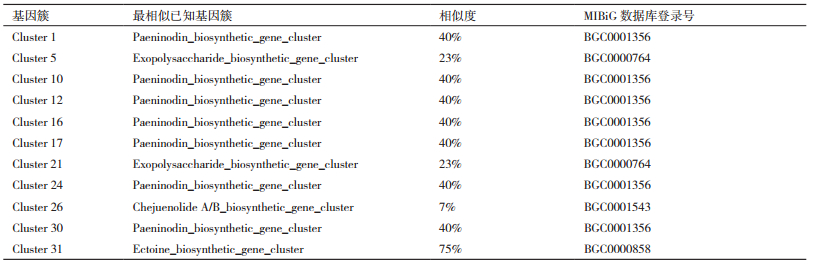
在上述基因簇中,基因簇1、5、10、12、16、17、21、24、26、30和31与已知基因簇具有一定同源性(表 3)。其中只有基因簇31与已知基因簇同源相似度较高,为75%,其他基因簇的同源相似度均低于40%。这说明,环状芽孢杆菌中的次级代谢产物可能与已知的次级代谢产物有所不同。
在GenBank数据库中,有9株环状芽孢杆菌菌株具有基因组数据,其中有7株只组装到contig水平。在本研究中,首先分析了9株菌的进化关系,发现9株菌在进化树上被归为了两个分支。我们又对9种环状芽孢杆菌基因组进行了的泛基因组和核心基因组分析,共鉴定出4 593个特有基因,其中菌株NCTC2610的基因最多(3 030个),而菌株NBRC 13626的特有基因最少(39个)。另外,共鉴定出3 622个基因家族组成核心基因组,占环状芽孢杆菌泛基因组的37.83%。赵永兵[27]对甲型副伤寒菌进行核心基因组分析发现,其核心基因组占泛基因组大小的87.5%;与其相比,环状芽孢杆菌在泛基因组组成上可能保守性较低。通过计算泛基因组、核心基因组和基因组数目之间的关系发现,随着基因组测序数量的增加,泛基因组随之增加,而核心基因组则逐渐减少。因此,我们可以推测环状芽孢杆菌的泛基因组是开放的。
通过次级代谢产物合成基因簇分析,9个环状芽孢杆菌基因组中共发现6类、32个次级代谢基因簇,重复出现最多的代谢通路是羊毛硫肽、套索肽和萜烯类化合物合成通路; 另外,有11个基因簇与已知基因簇具有一定同源性,其中基因簇31与已知基因簇同源相似度较高。这些结果说明,环状芽孢杆菌可能具有相似的代谢产物合成途径,最有可能的活性物质是羊毛硫肽、套索肽和萜烯类化合物。Letzel等[28]对211个已发表的厌氧细菌基因组进行了挖掘,发现25%以上的菌株具有翻译后修饰多肽合成基因簇(972个)。Zhang等[29]对830个已发表的放线菌基因组进行了挖掘,共发现1 163个类羊毛硫肽合成基因簇。Xin和Kuipers[30]对57个种328个已发表的芽孢杆菌目细菌基因组进行了挖掘,13类2 397个次级代谢合成通路。与这些类型的细菌相比,环状芽孢杆菌的次级代谢通路比较少,可能合成的新型物质可能也比较少。
4 结论本文通过对9个环状芽孢杆菌基因组进行分析,明确了其泛基因组含有9 572个基因家族,核心基因组含有3 622个基因家族,鉴定出4 593个特有基因; 通过次级代谢产物合成基因簇分析,9个环状芽孢杆菌基因组中共发现6类、32个次级代谢基因簇,重复出现最多的代谢通路是羊毛硫肽、套索肽和萜烯类化合物的合成通路。
| [1] |
Logan NA, Old DC, Dick HM. Isolation of Bacillus circulans from a wound infection[J]. Journal of Clinical Pathology, 1985, 38(7): 838. DOI:10.1136/jcp.38.7.838 |
| [2] |
Hemmati-Brivanlou A, Melton DA. Inhibition of activin receptor sig-naling promotes neuralization in Xenopus[J]. Cell, 1994, 77(2): 273-281. |
| [3] |
Watanabe T, Kobori K, Miyashita K, et al. Identification of glutamic acid 204 and aspartic acid 200 in chitinase A1 of Bacillus circulans WL-12 as essential residues for chitinase activity[J]. Journal of Biological Chemistry, 1993, 268(25): 18567-18572. |
| [4] |
Lawson CL, van Montfort R, Strokopytov B, et al. Nucleotide sequence and X-ray structure of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 in a maltose-dependent crystal form[J]. Journal of Molecular Biology, 1994, 236(2): 590-600. DOI:10.1006/jmbi.1994.1168 |
| [5] |
李才明, 黄敏, 顾正彪, 等. 来源于Bacillus circulan的重组β-CGT酶的分离纯化及其生化性质分析[J]. 食品与生物技术学报, 2018, 217(4): 30-38. |
| [6] |
Wakarchuk WW, Campbell RL, Sung WL, et al. Mutational and crystallographic analyses of the active site residues of the Bacillus circulans xylanase[J]. Protein Science, 1994, 3(3): 467-475. |
| [7] |
Yin H, Bultema JB, Dijkhuizen L, et al. Reaction kinetics and galactooligosaccharide product profiles of the β-galactosidases from Bacillus circulans, Kluyveromyces lactis and Aspergillus oryzae[J]. Food Chemistry, 2017, 225: 230-238. DOI:10.1016/j.foodchem.2017.01.030 |
| [8] |
孙德四, 王化军, 张强. 环状芽孢杆菌对铝土矿浸出分解行为的影响[J]. 中国有色金属学报, 2013, 23(4): 1119-1128. |
| [9] |
McLeod C. Circulin, an antibiotic from a member of the Bacillus circulans group:I. bacteriological Studies[J]. J Bacteriol, 1948, 56(6): 749-754. |
| [10] |
Dion HW, Woo PWK, Willmer NE, et al. Butirosin, a new aminog-lycosidic antibiotic complex:isolation and characterization[J]. Antimicrobial Agents and Chemotherapy, 1972, 2(2): 84-88. DOI:10.1128/AAC.2.2.84 |
| [11] |
He H, Shen B, Korshalla J, et al. Circulocins, new antibacterial lipopeptides from Bacillus circulans, J2154[J]. Tetrahedron, 2001, 57(7): 1189-1195. DOI:10.1016/S0040-4020(00)01135-2 |
| [12] |
Das P, Mukherjee S, Sen R. Antimicrobial potential of a lipopeptide biosurfactant derived from a marine Bacillus circulans[J]. Journal of Applied Microbiology, 2008, 104(6): 1675-1684. DOI:10.1111/j.1365-2672.2007.03701.x |
| [13] |
王美琴, 贺运春, 刘慧平, 等. 内生环状芽孢杆菌Jcxy8对番茄灰霉病的防病机制研究[J]. 中国生态农业学报, 2010, 18(1): 98-101. |
| [14] |
Tettelin H, Masignani V, Cieslewicz MJ, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae:implications for the microbial "pan-genome"[J]. Proceedings of the National Academy of Sciences, 2005, 102(39): 13950-13955. DOI:10.1073/pnas.0506758102 |
| [15] |
Yang X, Li Y, Zang J, et al. Analysis of pan-genome to identify the core genes and essential genes of Brucella spp.[J]. Molecular Genetics and Genomics, 2016, 291(2): 905-912. |
| [16] |
Vernikos G, Medini D, Riley DR, et al. Ten years of pan-genome analyses[J]. Current Opinion in Microbiology, 2015, 23: 148-154. DOI:10.1016/j.mib.2014.11.016 |
| [17] |
Mira A, Martín-Cuadrado AB, D'Auria G, et al. The bacterial pan-genome:a new paradigm in microbiology[J]. International Microbiology, 13(2): 45-57. |
| [18] |
胡彦婷, 安丽康, 尹淑丽, 等. 推定第四类羊毛硫素合成酶生物信息学分析[J]. 微生物学通报, 2016, 43(11): 2464-2472. |
| [19] |
Polter SJ, Caraballo AA, Lee YP, et al. Isolation, identification, whole-genome sequencing, and annotation of four Bacillus species, B. anthracis RIT375, B. circulans RIT379, B. altitudinis RIT380, and B. megaterium RIT381, from internal stem tissue of the insulin plant costus igneus[J]. Genome Announc, 2015, 3(4): e00847. |
| [20] |
Bertels F, Silander OK, Pachkov M, et al. Automated reconstruction of whole-genome phylogenies from short-sequence reads[J]. Molecular Biology and Evolution, 2014, 31(5): 1077-1088. DOI:10.1093/molbev/msu088 |
| [21] |
Kumar S, Stecher G, Li M, et al. MEGA X:molecular evolutionary genetics analysis across computing platforms[J]. Molecular Biology and Evolution, 2018, 35(6): 1547-1549. DOI:10.1093/molbev/msy096 |
| [22] |
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences[J]. Journal of Molecular Evolution, 1980, 16(2): 111-120. |
| [23] |
Chen X, Zhang Y, Zhang Z, et al. PGAweb:a web server for bacterial pan-genome analysis[J]. Frontiers in Microbiology, 2018, 9. |
| [24] |
Zhao Y, Wu J, Yang J, et al. PGAP:pan-genomes analysis pipeline[J]. Bioinformatics, 2011, 28(3): 416-418. |
| [25] |
Zhao Y, Jia X, Yang J, et al. PanGP:a tool for quickly analyzing bacterial pan-genome profile[J]. Bioinformatics, 2014, 30(9): 1297-1299. DOI:10.1093/bioinformatics/btu017 |
| [26] |
Blin K, Wolf T, Chevrette MG, et al. antiSMASH 4. 0-improveme-nts in chemistry prediction and gene cluster boundary identification[J]. Nucleic Acids Research, 2017, 45(W1): W36-W41. DOI:10.1093/nar/gkx319 |
| [27] |
赵永兵.泛基因组学分析方法开发及应用[D].北京: 中国科学院, 2014. http://cdmd.cnki.com.cn/Article/CDMD-80167-1015560078.htm
|
| [28] |
Letzel AC, Pidot SJ, Hertweck C. Genome mining for ribosomally synthesized and post-translationally modified peptides(RiPPs)in anaerobic bacteria[J]. BMC Genomics, 2014, 15(1): 983. DOI:10.1186/1471-2164-15-983 |
| [29] |
Zhang Q, Doroghazi JR, Zhao X, et al. Expanded natural product diversity revealed by analysis of lanthipeptide-like gene clusters in actinobacteria[J]. Applied and Environmental Microbiology, 2015, 81(13): 4339-4350. DOI:10.1128/AEM.00635-15 |
| [30] |
Xin Z, Kuipers OP. Identification and classification of known and putative antimicrobial compounds produced by a wide variety of Bacillales species[J]. BMC Genomics, 2016, 17(1): 882. DOI:10.1186/s12864-016-3224-y |