




2. 江南大学生物工程学院,无锡 214122
2. School of Biotechnology, Jiangnan University, Wuxi 214122
三羟基雄甾烯酮是德国先令公司推出的第四代女用口服避孕药有效成分屈螺酮的重要中间体[1-2]。自上市以来,“优思明”的销售额已占全球女用口服避孕药的首位,使得三羟基雄甾烯酮有着较大的市场需求[3]。传统的化学合成法是以醋酸去氢表雄酮作为底物来合成屈螺酮,需要反应步骤18步[4]。此合成路线反应条件苛刻,立体选择性差,产物收率较低,并且会产生大量的污染物[5]。随着微生物转化在甾体药物合成中的深入研究,具有将DHEA双羟化合成三羟基雄甾烯酮的菌株已被研究者从自然界中筛选获得,建立了生物-化学法合成屈螺酮的新工艺[6],其中关键几步采用微生物转化法[7]。与传统化学合成法相比,甾体生物转化法具有较高的立体选择性,转化条件温和、环境友好等优势[8],被誉为“绿色化学”[9-10]。目前,转化液中产物的分离提取通常是转化液经离心后,用亲水性有机溶剂萃取菌体,用非极性有机溶剂萃取上清液[11],这种提取分离方式与传统方法直接用乙酸乙酯萃取相比,能够提高产物提取率。粗品浓缩后经柱层析洗脱、浓缩,重结晶获得所需纯品。
本实验室从土壤中筛选一株亚麻刺盘孢ST-1(Colletotrichumlini ST-1)菌株,可以直接催化底物DHEA得到双羟化产物7α,15α-diOH-DHEA。本实验首先优化转化液预处理的方式和萃取条件来提高产物提取率,进而优化柱层析中洗脱剂种类和比例,确定绿色高效的纯化条件,从而建立最适的生物法制备三羟基雄甾烯酮的分离提取工艺。
1 材料与方法 1.1 材料 1.1.1 菌株亚麻刺盘孢ST-1(Colletotrichumlini ST-1),由江南大学实验室分离并保存。
1.1.2 培养基斜面培养基(g·L-1):马铃薯200,葡萄糖20,琼脂20;种子培养基(g·L-1):葡萄糖15,酵母粉15,豆饼粉10,玉米浆3;转化培养基(g·L-1):葡萄糖15,酵母粉15,玉米浆3。
1.1.3 主要试剂甲醇、乙醇、氯仿、丙酮、二氯甲烷、乙酸乙酯、石油醚、正己烷、乙腈(色谱纯)、层析用硅胶(200-300目)和D101大孔吸附树脂等试剂购自国药集团化学试剂有限公司,层析硅胶G薄板购自于武汉新技术开发有限公司,DHEA、7α,15α-diOH-DHEA购自浙江仙琚制药股份有限公司。
1.1.4 主要仪器DK-8D型电热恒温水浴锅(上海医用恒温设备厂),高功率超声波清洗器KQ-800KDE(昆山市超声仪器有限公司),旋转蒸发仪(上海亚荣有限公司),Nicolet iS10型红外光谱仪(美国赛默飞公司),柱层析色谱(美国GRACE公司),TSQ Quantum μLtra EMR型液质联用仪(美国赛默飞公司),U3000分析型液相色谱(美国戴安科技公司),Aduance Ⅲ全数字化核磁共振声波仪(瑞士布鲁克公司)。
1.2 方法 1.2.1 C.lini ST-1菌种的培养从斜面挑取适量的菌丝体,接种于100 mL新鲜种子培养基(500 mL锥形瓶)中,在30℃,220 r/min的摇床上培养48 h。将获得的一级种子液以10%(V/V)的接种量接入新鲜种子培养基中,在30℃,220 r/min的摇床上培养24 h,获得二级种子液。
1.2.2 C.lini ST-1菌种的转化二级种子液以10%(V/V)的接种量接入30 mL的转化培养基(250 mL锥形瓶)中,在30℃,220 r/min的摇床上培养24 h后投加10 g/L的DHEA,转化72 h。
1.2.3 产物7α,15α-diOH-DHEA的测定转化液的上清和菌丝体经萃取复溶后,通过高效液相色谱仪(戴安U3000)测定7α,15α-diOH-DHEA质量浓度。
7α,15α-diOH-DHEA提取率(%)= 7α,15α-diOH-DHEA的实际测得的含量(g)/7α,15α-diOH-DHEA的理论含量(g)×100%。
1.2.4 转化液的预处理(1) 加热处理:取100 mL转化液分别在20、40、60、80和100℃水浴锅中放置20 min,并用等体积的乙酸乙酯对转化液进行萃取。(2)调节pH值:取100 mL转化液,调节转化液的pH值,分别调节为3、5、7、9、11,并用等体积的乙酸乙酯对转化液进行萃取。(3)超声处理:取100 mL转化液,在超声功率分别为200、300、400、500和600 W下超声15 min,并用等体积的乙酸乙酯对转化液进行萃取。(4)化学处理:在100 mL转化液中加入1%-10%(V/V)乙醇-氢氧化钠溶液(2 mol/L),混合均匀后用等体积的乙酸乙酯对转化液进行萃取。[乙醇-氢氧化钠溶液配制为乙醇:氢氧化钠溶液=1: 1(V/V)]。
1.2.5 转化液的萃取转化液经离心处理后,上清液分别选择等体积的石油醚、正己烷、乙酸乙酯、二氯甲烷以及氯仿萃取2遍,沉淀则分别选择等体积的甲醇、乙醇和丙酮萃取2遍。合并两种萃取液,经浓缩及乙腈复溶后,经HPLC检测分析。
1.2.6 产物的分离与检测(1) 展开剂的选择:根据溶剂的体积比,选择二氯甲烷:甲醇=15: 1、10: 1、5: 1;氯仿:甲醇= 15: 1、10: 1、5: 1;氯仿:乙醇= 15: 1、10: 1、5: 1;二氯甲烷:乙醇= 15: 1、10: 1、5: 1作为展开剂,经薄层色谱分析,根据展开效果与展开时间,选择最佳的展开剂。(2)硅胶的处理:将700 mL体积的硅胶浸没于甲醇溶液中,超声处理20 min后,将含硅胶的溶液抽滤后,烘干硅胶。(3)样品的制备:用甲醇溶解所需的粗提物,与200-300目的硅胶混匀,利用旋转蒸发仪浓缩至细小颗粒,用于柱层析的分离[12]。(4)各组分的分离:选用二氯甲烷与乙醇作为流动相,流速设定为22 mL/min,按乙醇体积百分比进行梯度洗脱,程序为:1%-6%-7%-10%-100%。使用Grace RevelerisTM Flash色谱系统中的蒸发光散射检测器和80 g的硅胶柱对各组分检测与分离。根据薄层色谱分离效果及分离物质的Rf值,确定洗脱条件。(5)洗脱组分的检测:将不同时候出峰所对应的洗脱液收集后,用薄层色谱进行分析,将结果相同的洗脱液合并,并通过HPLC进行测定。
1.2.7 薄层层析(TLC)吸取5 μL粗提物在TLC板上点样,使用氯仿:甲醇=15: 1作为展开剂,在层析缸中展开10-16 min,吹干后用乙醇:浓硫酸= 1: 1的显色剂显色[13]。
1.2.8 高效液相分析(HPLC)粗提物复溶于乙腈中,用0.22 μm有机滤膜过滤后通过HPLC进行测定,使用安捷伦C18反相柱(4.6×250 mm,5 μm)。HPLC条件如下:检测波长206 nm,柱温30℃,流动相(乙腈:水=7: 3),进样量10 μL,流速0.5 mL/min[14]。
1.2.9 转化产物的结构鉴定(1) 红外光谱分析:采用KBr压片法制备压片,使用Nicolet iS10型红外光谱仪进行检测。(2)质谱分析:使用TSQ Quantum μLtra EMR型液质联用仪,模式:阳离子模式。(3)氢谱分析:使用Aduance Ⅲ全数字化核磁共振声波仪在25℃下检测,溶剂:氘代二甲基亚砜。
2 结果 2.1 转化液的预处理通过物理法和化学法对转化液进行预处理,改变细胞膜的通透性,使胞内产物更好的出胞,从而提高提取率。图 1为转化液预处理方式的比较结果,选取加热处理最佳条件60℃,转化液处理后最佳pH 8,超声处理最佳功率500 W,以及氢氧化钠-甲醇溶液添加量10%(V/V)。通过调节转化液pH的方式,对产物的提取效果作用不明显,调节温度和改变超声功率对产物提取效果作用较小,而化学处理法能够增大细胞壁的通透性,促进胞内物质释放,故添加氢氧化钠-甲醇溶液,处理效果最为明显。用等体积乙酸乙酯萃取转化液,获得的粗品用硅胶柱进行产物分离,以氯仿:甲醇=15: 1作为洗脱剂,再通过重结晶得到纯品。因此,在转化液中添加一定浓度的氢氧化钠-乙醇溶液,具有较好的效果。

|
| 图 1 预处理效果评估 |
为了提高化学处理法对转化液预处理的效果,优化了氢氧化钠-乙醇溶液的添加量,结果如图 2所示,添加4%(V/V)的氢氧化钠-乙醇溶液效果最好,产物提取率可达85.2%。

|
| 图 2 氢氧化钠-乙醇的添加量对提取率的影响 |
产物的提取率与萃取剂的种类、用量等因素有关。用相应的萃取剂分别等体积的萃取沉淀和上清各2遍,表 1是优化萃取剂种类的结果,根据提取产物相对百分比,结果显示分别选用乙酸乙酯和乙醇对上清和沉淀的萃取效果最好。

转化液经预处理后,8 000 r/min离心10 min。将上清和沉淀分别用不同体积比的乙酸乙酯和乙醇萃取。将萃取液分开浓缩后,通过柱层析和重结晶分别得到了上清和沉淀中产物纯品。结果(图 3)表明,用1.5倍体积的乙酸乙酯萃取上清,上清中产物的质量达0.16 g;用2倍体积的乙醇萃取沉淀,沉淀中产物的质量达0.67 g,继续增加试剂的用量,沉淀中产物的质量不再增加。为提高产物提取率,上清先后用1倍和0.5倍体积的乙酸乙酯萃取,而沉淀用等体积的乙醇萃取2遍。

|
| 图 3 萃取剂用量对上清和沉淀中产物产量的影响 |
氯仿、甲醇、乙醇和二氯甲烷是极性较为相似有机溶剂,从中选取两种溶剂,按不同体积比例混合作为的展开剂,并通过薄层色谱分析,从而选择最适的洗脱剂配比,氯仿:甲醇(V/V)= 5: 1、10: 1;二氯甲烷:甲醇(V/V)= 5: 1、10: 1;氯仿:乙醇(V/V)= 5: 1、10: 1,因其分离效果不佳,未放入图中,其余比例结果如图 4。通过薄层色谱分析,在展开相同的长度下,通过各点的Rf值和展开时间,确定最佳的展开剂,结果如表 2。

|
| 图 4 不同展开剂下的TLC效果图 (A)氯仿:甲醇=15: 1;(B)二氯甲烷:甲醇=15: 1;(C)氯仿:乙醇= 15: 1;(D)二氯甲烷:乙醇=15: 1;(E)二氯甲烷:乙醇=10: 1;(F)二氯甲烷:乙醇=5: 1 |
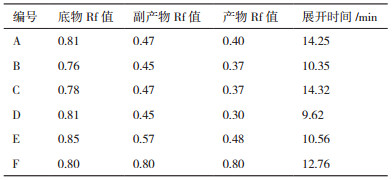
图 4-A-D中,底物、副产物和产物已经完全分开。表 2结果显示,根据各点的Rf值,当展开剂选择二氯甲烷:乙醇=15: 1时,展开速度最快,展开效果也较好。因此,该比例作为柱层析的洗脱剂,绿色高效又降低环境污染。
2.4 硅胶柱层析及产物的结晶所用菌株亚麻刺盘孢ST-1,其底物转化率已达97.1%,产物7α,15α-diOH-DHEA得率达80.1%。投加10 g/L的DHEA,500 mL的转化液经上述最优条件处理后,萃取后浓缩的粗品于外接硅胶柱上进行产物的分离纯化。图 5为硅胶柱上的分离效果图,每次上样量约为2.4 g,上样2次。洗脱速度为29 mL/min,A、B、C三种物质完全分离所需时间为42 min。

|
| 图 5 粗提物的分离 |
图 6为物质洗脱后在TLC上的结果。23 min时收集到A峰,31-32 min时收集到B峰,34-39 min时收集到C峰,从TLC的显色结果来看,产物、副产物与底物已被成功的分开。

|
| 图 6 不同物质的TLC分析 |
由于产物和副产物极性较为接近,在分离过程中,会有少量的副产物混入。为了提高产物纯度,将收集到的产物洗脱峰浓缩成粉末后,用乙酸乙酯复溶,并水浴加热,使溶液达到饱和状态,在室温下放置1 h后,再移入-20℃冰箱中过夜。将析出的晶体烘干称重,共计4.15 g即为所要产物其优化的提取分离路线,见图 7。

|
| 图 7 三羟基雄甾烯酮提取分离流程图 |
用乙腈复溶结晶后的样品C,通过HPLC分析(图 8),结晶后的样品C有单一的吸收峰,且与标准品7α,15α-diOH-DHEA的出峰时间相同,初步判定样品C为产物7α,15α-diOH-DHEA,经计算纯度为98.1%。

|
| 图 8 物质C的纯度分析 |
经过对比分析,纯化后得到的物质C与标准品7α,15α-diOH-DHEA的特征吸收峰一致,表现为在1 630 cm-1处的C=C伸缩振动峰;在1 720 cm-1处的C=O伸缩振动峰;在2 900 cm-1处的R-CH3伸缩振动峰;在3 450 cm-1处的-OH伸缩振动峰(图 9)。

|
| 图 9 物质C的红外光谱 |
纯化得到物质C在质谱图中有一个明显的峰,MS(ESI)m/z[M+K]+为359,所以物质C的分子量为320,与7α,15α-diOH-DHEA(C19H28O4)的分子量相同(图 10)。

|
| 图 10 物质C的质谱图 |
将纯化得到物质C的氢谱与7α,15α-diOH-DHEA标准品的氢谱进行比较,出峰的位置和位移基本相同。1H-NMR图谱,化学位移情况:1H NMR(400 MHz,DMSO)δ5.95(d,J = 4.1 Hz,1H),5.41(d,J = 4.8 Hz,1H),4.64(d,J = 4.3 Hz,1H),4.32(d,J = 4.5 Hz,1H),4.29-4.19(m,1H),4.11(d,J = 3.4 Hz,1H),2.89(dd,J = 18.9,7.6 Hz,1H),2.31-1.99(m,2H),1.92(dd,J = 19.1,6.4 Hz,1H),1.81-1.54(m,7H),1.47-1.31(m,2H),1.25(dq,J =11.6,3.9 Hz,2H),0.99-0.92(m,1H),0.89(s,3H),0.81(s,3H)。将纯化产物与Tatyana G等研究所得的7α,15α-OH-DHEA的1H-NMR图谱比对也基本一致,因此物质C的化学结构与7α,15α-diOH-DHEA一致(图 11)。

|
| 图 11 物质C的核磁共振氢谱 |
7α,15α-diOH-DHEA是合成甾体药物屈螺酮的关键中间体,作为“优思明”的主要成分,屈螺酮不仅具有显著的避孕效果,还能有效控制体重,稳定情绪[15],改善经前症状等作用[16],具有广阔的市场前景。生物化转化DHEA生成7α,15α-diOH-DHEA是目前工业上的主要合成方法,而目的产物的分离提取是决定生产成本的关键因素之一,因此建立一种绿色高效的分离提取工艺意义重大。
转化液的预处理,可以使胞内的产物更好的释放到胞外,一般有物理法、化学法和酶法。酶法处理具有操作温和,选择性强,但缺点是酶的成本较高,极大限度的限制了大规模的生产。物理法通过改变转化液温度、pH及超声破碎等方法来提高细胞内物质的释放。但转化液在物理法作用下产物提取率不佳,选用化学处理法即氢氧化钠-乙醇溶液处理转化液,通过增大细胞壁的通透性来提高提取率。此外,转化液的直接萃取需要使用大量的有机溶剂[17],而将转化液离心后得到的上清和沉淀,分别用亲水性和疏水性有机溶剂萃取,可提高提取率。
硅胶柱层析分离甾体类物质,通常使用甲醇和氯仿作为洗脱剂,但因分离过程中洗脱剂用量较大,且氯仿和甲醇都具有毒性。因此,使用与其极性相似的二氯甲烷和乙醇来替代氯仿和和甲醇,选用二氯甲烷和乙醇作为洗脱剂后,其洗脱效果较佳,更加绿色高效,减少对环境的污染。
目前,本实验室筛选得到的C.lini ST-1,与已报道具有将DHEA双羟化成7α,15α-diOH-DHEA的菌株相比,转化率较高[18]。通过对转化液提取和粗品分离纯化工艺的优化,7α,15α-diOH-DHEA的收率达91.4%,较文献报道有所提高[11],其优化的提取分离路线见图 11,将其应用于屈螺酮的生产,具有重大的市场价值。
4 结论产物7α,15α-diOH-DHEA分离纯化过程中,通过优化转化液预处理的方式和萃取条件,确定最终方案为,先将4%(V/V)的氢氧化钠-乙醇溶液添加到转化液中,充分振荡,经离心后分别用2倍样品体积的乙酸乙酯和1.5倍样品体积的乙醇萃取上清和沉淀。洗脱剂为二氯甲烷:乙醇= 15: 1的混合溶液,经梯度洗脱,底物、副产物和产物3种物质被成功分离。分别用红外光谱、质谱和氢谱对产物结构进行验证,确定产物为7α,15α-diOH-DHEA。虽然纯化中收率有所损失,但纯化收率仍达91.4%。
| [1] |
刘丰良. 屈螺酮中间体的合成新方法及其机理研究[D]. 武汉: 中南大学, 2005.
|
| [2] |
林晓辉, 章平荣, 柯贤炳, 等. 屈螺酮的合成工艺研究[J]. 科协论坛, 2007(6): 33-34. DOI:10.3969/j.issn.1007-3973.2007.06.021 |
| [3] |
韩广甸, 刘宏斌. 新型女用口服避孕药屈螺酮的合成进展[J]. 中国药物化学杂志, 2010(2): 146-154. |
| [4] |
何明华, 廖清江. 曲螺酮的合成[J]. 中国新药杂志, 2006, 15(20): 1756-1759. DOI:10.3321/j.issn:1003-3734.2006.20.015 |
| [5] |
吴燕. 亚麻刺盘孢ST-1转化去氢表雄酮合成三羟基雄甾烯酮的研究[D]. 无锡: 江南大学, 2015.
|
| [6] |
Petzoldt K, Laurent H, Wiechert R. A novel synthetic route to the aldosterone-antagonist spirorenone[J]. Angewandte Chemie International Edition, 1983, 22(5): 406-407. |
| [7] |
Donova MV, Egorova OV. Microbial steroid transformations:current state and prospects[J]. Applied Microbiology and Biotechnology, 2012, 94(6): 1423-1447. DOI:10.1007/s00253-012-4078-0 |
| [8] |
许正宏, 吴燕, 李会, 等. 甾体生物转化技术研究的现状与进展[J]. 生物加工过程, 2013, 11(2): 30-36. DOI:10.3969/j.issn.1672-3678.2013.02.005 |
| [9] |
Donova MV. Transformation of steroids by actinobacteria:a review[J]. Prikladnaia Biokhimiia Mikrobiologiia, 2007, 43(1): 5-18. |
| [10] |
ČrešnarB, ŽakeljMavričM. Aspects of the steroid response in fungi[J]. Chemico-Biological Interactions, 2009, 178(1-3): 303-309. |
| [11] |
张琴, 胡海峰, 陶荣盛. 7α, 15α-二羟基雄烯醇酮的提取纯化方法: 中国, CN101182565[P]. 2008-05-21.
|
| [12] |
付珍珍. 生物转化制备三羟基雄甾烯酮赤霉菌的筛选及工艺优化[D]. 无锡: 江南大学, 2014.
|
| [13] |
尚珂, 胡海泉, 朱宝泉. 微生物转化法合成7α, 15α-二羟基雄烯醇酮[J]. 中国医学生物技术应用, 2004(2): 30-34. |
| [14] |
李传鹏, 李会, 吴燕, 等. 复合诱变选育高效转化DHEA为7α, 15α-diOH-DHEA的菌株及其转化工艺优化[J]. 生物工程学报, 2014, 30(1): 147-156. |
| [15] |
周静, 雷贞武. 新型短效复方口服避孕药——屈螺酮炔雌醇片[J]. 中国计划生育学杂志, 2009, 17(6): 371-373. DOI:10.3969/j.issn.1004-8189.2009.06.023 |
| [16] |
Mansour D. Experiences with Yasmin:the acceptability of a novel oral contraceptive and its effect on well-being[J]. Eur J Contracept Reprod Health Care, 2002, 7(Suppl 3): 35-41. |
| [17] |
陈怡倩, 胡海峰, 朱宝泉. 微生物转化合成抗真菌活性化合物SIPI-9AAD[C]//全国抗生素. 2009: 448-452.
|
| [18] |
Romano A, Romano D, Ragg E, et al. Steroid hydroxylations with Botryodiplodia malorum and Colletotrichum lini[J]. Steroids, 2006, 71(6): 429-434. DOI:10.1016/j.steroids.2006.01.014 |