




亚磷酸盐脱氢酶[Phosphite Dehydrogenase(PTDH),EC 1.20.1.1]以NAD+作为辅酶,催化亚磷酸盐氧化为正磷酸盐,同时伴随NADH的产生[1]。该酶属于D-2-羟酸脱氢酶家族,含有3个保守的关键催化残基(Arg237、His292和Glu266)[2]以及辅酶NAD+的结合基序GxGxxG(17x)E[1]。亚磷酸盐脱氢酶的专一性极强,只能利用亚磷酸盐作为底物;而且它也通常只利用NAD+作为辅酶,对NADP+的亲和力极低[3]。
亚磷酸盐脱氢酶对底物的Km值很小,它氧化亚磷酸盐反应过程的平衡常数估计为1011,反应基本是不可逆的,而且正磷酸盐产物在高浓度下也不产生抑制效应[1, 3],这些特性再加上相对低廉的亚磷酸盐底物决定了它在辅酶再生系统中起着重要作用[1, 4]。而且,热稳定性的、对NADP+也具高亲和力的突变体的创建更是进一步拓展了该酶的应用范围[5-6]。
另外,亚磷酸盐脱氢酶能够将植物和大多数微生物不能代谢利用的亚磷酸盐氧化成为可代谢利用的正磷酸盐,这一转化过程可以赋予植物利用亚磷酸盐作为磷源的能力,以应对潜在的可用性磷资源短缺[7]和不断恶化的磷污染生态环境问题[8]。同时,伴随催化反应再生出来的还原型NADH/NADPH还是生物体大多数氧化还原酶所不可或缺的。而且,正是因为植物自身不能代谢利用亚磷酸盐以及后者反而抑制植物的生长发育[9],亚磷酸盐脱氢酶已被用作一种新型植物转基因选择标记和杂草控制系统,并在拟南芥[10-11]、烟草[10-11]、棉花[12]、玉米[13]和水稻[14]等植物中得到检验。这些优点无疑赋予了亚磷酸脱氢酶在转基因作物研究领域也有着广阔的应用前景[15]。
亚磷酸盐脱氢酶只存在于少数类细菌中[16],目前已从某些特定分离株中克隆了其基因[3, 17-19],其中就包括假单胞菌。虽然这类细菌在土壤中普遍存在,但仅靠它们将亚磷酸盐转化为正磷酸盐的量不足以维持植物生长发育所需的磷量,也不能消除用亚磷酸盐作为磷肥对植物的抑制作用。因此,要想亚磷酸盐成为一种真正有效磷肥,必须向目标植物导入亚磷酸盐脱氢酶基因并使之具备转化利用能力。
本研究首先直接从土壤宏基因组中扩增假单胞菌的亚磷酸盐脱氢酶基因PsPtx,然后分析它在大肠杆菌中的重组表达并检验重组蛋白PsPtx纯化后的酶活性,以便于其后续用于植物转基因研究。
1 材料与方法 1.1 材料大肠杆菌菌株DH5α、BL21(DE3)和载体pET32a(+)均为本实验室保存。植物根际土壤(根际地面下5 cm处潮湿土壤)采自校园。土壤基因组提取试剂盒、质粒提取试剂盒、琼脂糖凝胶回收试剂盒购于北京Tiangen生化科技有限公司;DNA及蛋白分子量标准以及DNA溶液纯化试剂盒等购于北京Transgene生物科技有限公司;Phanta Max Super-Fidelity DNA聚合酶、2× Taq Plus Master Mix购于南京诺唯赞生物科技有限公司;Pfu DNA聚合酶、T4 DNA连接酶和限制性内切酶NdeI、SacI等购于Thermo Scientific Fermentas公司;其它生化试剂均为国产分析纯。利用Vector NTI Advance 11软件设计引物(表 1),引物合成和DNA测序均由北京华大基因公司完成。
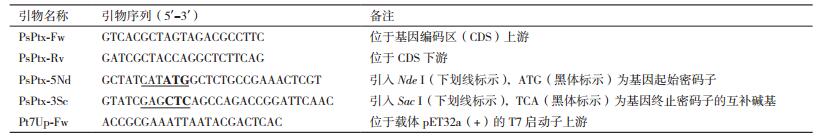
参照NCBI GenBank中已有的施氏假单胞菌(Psuedomonas stutzeri)WM88的PTDH基因PtxD序列(登录号AF061070)设计两对引物(PsPtx-Fw/PsPtx-Rv、PsPtx-5Nd/PsPtx-3Sc),从土壤中扩增和克隆亚磷酸盐脱氢酶基因PsPtx。参照Tiangen公司的土壤基因组DNA提取试剂盒说明书提取新采集土壤的宏基因组DNA。取1 μL作模板,使用引物PsPtx-Fw、PsPtx-Rv和Phanta Max Super-Fidelity DNA聚合酶进行第一轮PCR,程序为:95℃ 3 min;(95℃ 15 s,57℃ 15 s,72℃ 1 min)× 35;72℃ 10 min。然后,将第一轮产物稀释10倍,取1 μL作为模板,使用引物PsPtx-5Nd、PsPtx-3Sc和Pfu DNA聚合酶进行第二轮PCR,程序为:95℃ 5 min;(94℃ 30 s,47℃ 30 s,72℃ 2.5 min)×1;(94℃ 30 s,62℃ 30 s,72℃ 2.5 min)×30;72℃ 10 min。PsPtx PCR产物纯化后用Nde I和Sac I进行双酶切,再与经同样双酶切并回收的载体pET32a(+)大片段连接,构建成其大肠杆菌表达载体pET(PsPtx)。重组克隆用引物Pt7Up-Fw、PsPtx-3Sc通过菌落PCR鉴定,并使用载体上的T7启动子通用引物和T7终止子通用引物进行双向测序。
1.2.2 PsPtx的生物信息学分析DNA和蛋白质序列分析采用Vector NTI Advance 11软件。保守结构域的预测利用NCBI(http://www.ncbi.nlm.nih.gov)的在线CCD工具。另外,分别利用NCBI的BlastN和BlastP工具进行核酸和蛋白质序列的比对分析,并在比对结果的基础上进一步通过邻近法建立系统进化树。
1.2.3 PsPtx基因的诱导表达将重组质粒pET(PsPtx)转化大肠杆菌BL21(DE3),从平板上挑取单菌落于LB液体培养基(含100 μg/mL氨苄青霉素),37℃振荡培养过夜;按1:100的比例转接并于37℃振荡培养至OD600为0.6,然后加入终浓度为0.5 mmol/L的IPTG,于25℃诱导培养过夜或者37℃诱导培养4 h。将菌液离心并重悬于PBS缓冲液(pH 7.4)进行超声破碎。取16 μL细菌裂解物于14 000 r/min离心5 min;全部上清作为样品S,沉淀用16 μL PBS缓冲液重悬后作为样品P,同时取16 μL细菌裂解物作为样品T。另外,在加IPTG前取200 μL菌液,离心后菌体用16 μL PBS缓冲液重悬,作为未诱导样品(UI)。所有样品加4 μL 5×蛋白上样缓冲液,混匀后煮沸5 min,然后用12% SDS-PAGE进行电泳分析。通过比较诱导前后的蛋白电泳条带判断PsPtx基因在大肠杆菌中的表达情况。
1.2.4 重组蛋白PsPtx的纯化及酶活测定将pET(PsPtx)诱导表达菌的超声裂解物离心,取上清,然后采用GE Healthcare公司的HisTrap FF crude柱子和参照产品使用说明书进行重组蛋白PsPtx的纯化,并用12% SDS-PAGE检测蛋白纯化结果。利用BCA法对蛋白样品进行定量。将975 μL 20 mmol/L MOPS溶液(含6 mmol/L Na2PO3.5H2O、1.2 mmol/L NAD+)与25 μL纯化的PsPtx酶液在25℃水浴锅中保温10 min后,于分光光度计中测定OD340处的吸光值变化,根据酶液活性计算公式X(U/mL)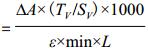
土壤中假单胞菌种类丰富,本工作采集根际地面下5 cm处潮湿土壤并提取其宏基因组DNA。取1 μL作为模板,用引物PsPtx-Fw和PsPtx-Rv进行第一轮PCR扩增,取5 μL进行电泳检测,结果显示目的基因扩增产物PsPtx-1大小正确(约1.1 kb),但条带亮度较弱(图 1-A)。将第一轮PCR产物PsPtx-1稀释10倍,取1μL作为模板,利用引物PsPtx-5Nd和PsPtx-3Sc进行第二轮PCR(巢式),取5 μL进行电泳检测,结果获得了非常明亮、特异性好、大小正确(约1 kb)的目的基因扩增产物PsPtx(图 1-B)。由于引物PsPtx-5Nd、PsPtx-3Sc的5’末端分别引入了酶切位点Nde I和Sac I,因此第二轮PCR产物PsPtx通过Nde I、Sac I双酶切,直接克隆到了大肠杆菌表达载体pET32a(+)上,菌落PCR(使用引物Pt7Up-Fw和PsPtx-3Sc)条带明亮、大小正确(图 1-C),表明获得了土壤PsPtx基因的大肠杆菌重组表达载体pET(PsPtx)。

|
| 图 1 PsPtx基因的克隆 1:第一轮PCR产物PsPtx-1;2:第二轮PCR产物PsPtx;3-5:重组克隆pET(PsPtx)的菌落PCR鉴定;M:100 bp DNA ladder plus;箭头标示目的DNA条带 |
利用T7启动子和终止子引物对所构建载体pET(PsPtx)进行双向测序,结果表明扩增的PsPtx基因编码区(CDS)长1 011 bp。其推导蛋白PsPtx由336个氨基酸组成,理论分子量大小为36.5 kD,含有辅酶NAD+的结合基序GxGxxG(17x)E,并具有保守的催化残基R、E、H(图 2)。另外,通过NCBI的CCD工具分析推测,PsPtx蛋白属于亚磷酸盐脱氢酶(PTDH,cd12157),含有NAD+的结合位点并以二聚体形式起作用。

|
| 图 2 PsPtx基因的核苷酸序列及其推导的氨基酸序列 下划线:辅酶NAD+的结合基序GxGxxG(17x)E;黑体阴影字母:结合基序中的关键残基;黑体带框字母:保守的催化残基 |
通过NCBI的BlastN工具将PsPtx CDS序列与核酸数据库进行比对分析,遴选适合以下标准(Cutoff:Query cover,100%;E value,0;Ident,98%-100%)的比对项,并通过邻近法建立了其系统进化树。由图 3可以看出,本工作从土壤宏基因组中扩增到的PsPtx基因应该来源于假单胞菌属(Pseudomonas sp.),但无法确定其种类。而且,它与引物设计的参照序列——施氏假单胞菌PtxD(登录号AF061070)也不完全相同。

|
| 图 3 PsPtx基因的系统进化树分析 Klebsiella:克雷白氏杆菌;Pseudomonas:假单胞菌;Salmonella:沙门氏菌;Serratia:沙雷氏菌;Thioalkalivibrio:巯碱弧菌 |
进一步通过NCBI的BlastP工具将PsPtx基因的推导蛋白序列与蛋白数据库进行比对分析,遴选适合以下标准(Cutoff:Query cover,80%-100%,E value,0;Ident,95%-100%)的比对项,并通过邻近法建立了其系统进化树。由图 4可以看出,本工作中PsPtx基因的编码蛋白PsPtx与数据库中任一从确定生物体中鉴定或推导出的蛋白都不一样,当然与引物设计的参照序列(施氏假单胞菌PtxD,登录号AF061070)的对应编码蛋白(登录号4NU5_A)也存在差异。但是,它与同样从宏基因组(主体菌为假单胞菌)中推导出的蛋白序列(登录号WP_003118429)却完全相同。

|
| 图 4 PsPtx编码蛋白的系统进化树分析 Klebsiella:克雷白氏杆菌;Pseudomonas:假单胞菌;Thioalkalivibrio:巯碱弧菌 |
将载体pET(PsPtx)转入大肠杆菌BL21(DE3)中,25℃经IPTG诱导表达过夜或37℃诱导表达4 h,然后进行SDS-PAGE分析。由图 5可以看出,无论是25℃还是37℃,与未诱导(UI)样品相比,诱导后总蛋白(T)、沉淀(P)和上清(S)样品在蛋白分子量标准40 kD条带下面多出一条明亮的条带(箭头标示),而且与PsPtx蛋白的理论分子量大小(36.5 kD)一致,说明两个温度下重组载体pET(PsPtx)在大肠杆菌中都获得了高效表达。但是,重组PsPtx蛋白在37℃下诱导表达时几乎不可溶并聚集在包涵体沉淀中,而在25℃诱导表达时可溶性则显著增加,符合低温诱导可提高大肠杆菌重组表达蛋白可溶性这一经典结论。

|
| 图 5 PsPtx基因在大肠杆菌BL21(DE3)中的重组表达 M:蛋白质分子量标准;UI:未诱导细菌;T:细菌裂解总蛋白;P:“T”的沉淀;S:“T”的上清;箭头标示重组表达蛋白PsPtx |
由于25℃诱导表达PsPtx蛋白的可溶性较高(图 5),因而可进行蛋白纯化及酶活测定分析。重新在25 mL pET(PsPtx)重组菌液中加IPTG诱导过夜,取14 mL表达菌体重悬于PBS溶液并超声破碎,离心后将上清液经HisTrap FF crude柱子纯化,然后将纯化过程各步的蛋白样品一起进行SDS-PAGE分析。结果(图 6)同样显示25℃诱导表达时重组蛋白PsPtx不但量高而且可溶性较好。另外,重组蛋白PsPtx与His-标签亲和柱结合效果佳,在上样流出液中几乎检测不到残存条带。通过100 mmol/L咪唑和200 mmol/L咪唑溶液洗脱都可获得单一的条带(分子量约为36.5 kD),但100 mmol/L咪唑浓度下洗脱不够彻底,而在200 mmol/L咪唑浓度下洗脱效果最好、纯化收率也最高。

|
| 图 6 大肠杆菌重组表达蛋白PsPtx的纯化 M:蛋白质分子量标准;UI:未诱导细菌;T:细菌裂解总蛋白;P:“T”的沉淀;S:“T”的上清;1:上样流出液;2:100 mmol/L咪唑出液;3:200 mmol/L咪唑出液;箭头标示重组表达蛋白PsPtx |
将25 μL纯化后的PsPtx酶液与其底物(NAD+、Na2PO3)在25℃保温反应后,通过分光光度法测定其OD340吸光值的变化,以此计算出其酶活性为5.4 U/mL。同时,通过BCA法测定纯化后PsPtx酶液的蛋白含量约为1.44 mg/mL,最终计算出重组PsPtx蛋白的酶比活为3.75 U/mg。
3 讨论亚磷酸盐脱氢酶因其生化特性及与植物磷代谢利用之间的关系,使得它在生物催化的辅酶再生方面和植物转基因领域具有广阔的应用潜能[1, 3, 10-15],并日益引起越来越多的研究关注。本工作从土壤中获取假单胞菌亚磷酸盐脱氢酶基因PsPtx并验证它在大肠杆菌重组表达蛋白的酶活性,也主要是为了它的进一步应用打下前期基础。
目前,亚磷酸盐脱氢酶基因都是从包括假单胞菌在内的特定分离菌株中得到克隆[3, 17-19],其中某些菌株因其特定生长环境并不容易获取、分离和培养。假单胞菌属在多数土壤中普遍存在,因而土壤实际上是亚磷酸盐脱氢酶基因获取的最简捷来源。本研究采用两轮PCR就首次成功从新鲜提取的土壤宏基因组中扩增得到亚磷酸盐脱氢酶基因PsPtx(图 1),尽管其首轮PCR产物很弱,但这对从任何宏基因组中扩增目的基因来说是个普遍现象,毕竟特定基因的模板DNA在整个宏基因组中的含量是极其低微的。总体上看,这种不需要分离特定生物体(特别是一些无法培养的菌株)就可以从中获得目的基因的策略是值得推广的,我们在之前的研究中也采用同样方法分别从蓝藻水体和人粪便宏基因组中成功分离到蓝藻橙色类胡萝卜素蛋白和甲基戊糖梭菌膜整合焦磷酸酶的基因[20-21]。
生物信息学分析显示本工作克隆的PsPtx基因含有一个完整的编码区,其推导蛋白(336 aa)属于PTDH家族成员,含有保守的催化功能残基(R、E和H)和辅酶NAD+的结合基序GxGxxG(17x)E(图 2)。通过高遴选标准的序列比对及其基础上的系统进化树分析,结果显示PsPtx基因来源于一无法确定种名的土壤假单胞菌,而且与参照基因施氏假单胞菌PtxD虽然在核酸或是蛋白序列上同源性很高,但在进化关系上却相对不太靠近(图 3,图 4)。
另外,本研究还发现,无论25℃还是37℃通过IPTG诱导,PsPtx基因在大肠杆菌BL21(DE3)中都获得了高水平表达,但重组蛋白在37℃诱导时几乎不可溶而在25℃诱导时可溶性则显著增加(图 5)。该结果说明重组PsPtx是一个易聚集和热不稳定蛋白,这与其它有关施氏假单胞菌PtxD重组表达的研究报道[5, 18]在很大程度上是相一致的。
最后,我们还验证了重组PsPtx蛋白在纯化后的酶活性。25℃下大肠杆菌诱导表达,重组蛋白PsPtx经His标签亲和层析,可在200 mmol/L咪唑浓度下几乎完全被洗脱(图 6)。通过分光光度法测定产物NADH的OD340吸光值变化来测定PsPtx纯化酶液的酶活性,数据显示本工作中PsPtx重组蛋白的酶比活性为3.75 U/mg,非常接近于以前其它相关研究的结果[3, 18-19]。
总之,本工作从土壤宏基因组中分离获得了活性验证过的假单胞菌亚磷酸盐脱氢酶基因PsPtx,为其后续的植物转基因应用研究提供了有力支撑。
4 结论本研究直接从根际土壤提取的宏基因组中通过两轮PCR扩增得到亚磷酸盐脱氢酶全长基因PsPtx。生物信息学分析显示该基因含有一个完整的编码区(长1 011 bp),其推导蛋白(336 aa)属于PTDH家族成员,含有辅酶NAD+的结合基序GxGxxG(17x)E和保守的催化残基(R、E、H)。系统进化树分析显示PsPtx基因来源于一无法确定种名的土壤假单胞菌,且与施氏假单胞菌在进化关系上还相对不太靠近。另外,PsPtx基因经诱导后可在大肠杆菌中获得高效表达,重组蛋白在37℃诱导时几乎不可溶但在25℃诱导时可溶性显著增加。此外,通过His标签亲和层析纯化了PsPtx重组蛋白并进行了酶活测定,其比活性为3.75 U/mg。
| [1] |
Relyea HA, van der Donk WA. Mechanism and applications of phosphite dehydrogenase[J]. Bioorg Chem, 2005, 33(3): 171-189. DOI:10.1016/j.bioorg.2005.01.003 |
| [2] |
Woodyer R, Wheatley JL, Relyea HA, et al. Site-directed mutagenesis of active site residues of phosphite dehydrogenase[J]. Biochemistry, 2005, 44(12): 4765-4774. DOI:10.1021/bi047868c |
| [3] |
Costas AM, White AK, Metcalf WW. Purification and characterization of a novel phosphorus-oxidizing enzyme from Pseudomonas stutzeri WM88[J]. J Biol Chem, 2001, 276(20): 17429-17436. DOI:10.1074/jbc.M011764200 |
| [4] |
Vrtis JM, White AK, Metcalf WW, et al. Phosphite dehydrogenase:a versatile cofactor-regeneration enzyme[J]. Angew Chem Int Ed Engl, 2002, 41(17): 3257-3259. DOI:10.1002/1521-3773(20020902)41:17<>1.0.CO;2-C |
| [5] |
McLachlan MJ, Johannes TW, Zhao H. Further improvement of phosphite dehydrogenase thermostability by saturation mutagenesis[J]. Biotechnol Bioeng, 2008, 99(2): 268-274. DOI:10.1002/(ISSN)1097-0290 |
| [6] |
Johannes TW, Woodyer RD, Zhao H. Efficient regeneration of NADPH using an engineered phosphite dehydrogenase[J]. Biotechnol Bioeng, 2007, 96(1): 18-26. DOI:10.1002/(ISSN)1097-0290 |
| [7] |
MacDonald GK, Bennett EM, Potter PA, et al. Agronomic phosphorus imbalances across the world' s croplands[J]. Proc Natl Acad Sci USA, 2011, 108(7): 3086-3091. DOI:10.1073/pnas.1010808108 |
| [8] |
Carpenter SR. Phosphorus control is critical to mitigating eutrophication[J]. Proc Natl Acad Sci USA, 2008, 105(32): 11039-11040. DOI:10.1073/pnas.0806112105 |
| [9] |
Ratjen AM, Gerendás J. A critical assessment of the suitability of phosphite as a source of phosphorus[J]. J Plant Nutr Soil Sci, 2009, 172: 821-828. DOI:10.1002/jpln.v172:6 |
| [10] |
López-Arredondo DL, Herrera-Estrella L. Engineering phosphorus metabolism in plants to produce a dual fertilization and weed control system[J]. Nat Biotechnol, 2012, 30(9): 889-893. DOI:10.1038/nbt.2346 |
| [11] |
López-Arredondo DL, Herrera-Estrella L. A novel dominant selectable system for the selection of transgenic plants under in vitro and greenhouse conditions based on phosphite metabolism[J]. Plant Biotechnol J, 2013, 11(4): 516-525. DOI:10.1111/pbi.2013.11.issue-4 |
| [12] |
Pandeya D, Campbell LM, Nunes E, et al. ptxD gene in combination with phosphite serves as a highly effective selection system to generate transgenic cotton(Gossypium hirsutum L.)[J]. Plant Mol Biol, 2017, 95(6): 567-577. DOI:10.1007/s11103-017-0670-0 |
| [13] |
Nahampun HN, López-Arredondo D, Xu X, et al. Assessment of ptxD gene as an alternative selectable marker for Agrobacterium-mediated maize transformation[J]. Plant Cell Rep, 2016, 35(5): 1121-1132. DOI:10.1007/s00299-016-1942-x |
| [14] |
Manna M, Achary VM, Islam T, et al. The development of a phosphite-mediated fertilization and weed control system for rice[J]. Sci Rep, 2016, 6: 24941. DOI:10.1038/srep24941 |
| [15] |
Achary VMM, Ram B, Manna M, et al. Phosphite:a novel P fertilizer for weed management and pathogen control[J]. Plant Biotechnol J, 2017, 15(12): 1493-1508. DOI:10.1111/pbi.2017.15.issue-12 |
| [16] |
Figueroa IA, Coates JD. Microbial phosphite oxidation and its potential role in the global phosphorus and carbon cycles[J]. Adv Appl Microbiol, 2017, 98: 93-117. DOI:10.1016/bs.aambs.2016.09.004 |
| [17] |
Simeonova DD, Wilson MM, Metcalf WW, et al. Identification and heterologous expression of genes involved in anaerobic dissimilatory phosphite oxidation by Desulfotignum phosphitoxidans[J]. J Bacteriol, 2010, 192(19): 5237-5244. DOI:10.1128/JB.00541-10 |
| [18] |
Hirota R, Yamane ST, Fujibuchi T, et al. Isolation and characterization of a soluble and thermostable phosphite dehydrogenase from Ralstonia sp. strain 4506[J]. J Biosci Bioeng, 2012, 113(4): 445-450. DOI:10.1016/j.jbiosc.2011.11.027 |
| [19] |
Liu DF, Ding HT, Du YQ, et al. Cloning, expression, and characterization of a wide-pH-range stable phosphite dehydrogenase from Pseudomonas sp. K in Escherichia coli[J]. Appl Biochem Biotechnol, 2012, 166(5): 1301-1313. DOI:10.1007/s12010-011-9518-2 |
| [20] |
刘畅, 罗著, 张梦如, 等. 蓝藻橙色类胡萝卜素蛋白的基因克隆及在大肠杆菌中的异源表达和功能分析[J]. 生物技术通报, 2016, 32(7): 138-145. |
| [21] |
刘延娟, 杨玉梅, 刘娴, 等. 甲基戊糖梭菌膜整合焦磷酸酶提高转基因烟草的抗旱性[J]. 生物技术通报, 2017, 33(6): 81-88. |