




杂交现象在高等生物的进化过程中扮演着重要角色。研究表明由于不完全的地理隔离、生殖隔离,物种形成后仍然会存在不同程度的基因交流,有观点认为大约25%的植物、10%的动物曾经发生过杂交的现象[1]。相对于基因突变而言,杂交能够快速积累遗传变异,有利于增加群体的遗传多样性,促进新物种的形成[2];其次,杂交有利于生成高度侵入性的基因型[3-4];最后,因杂交过程而产生的遗传同化作用还可能引起物种灭绝[5-7]。从基因组学出发识别古老和近期的杂交事件,有助于深入理解物种之间的系统发育关系[8],为研究杂交优势[9-10]提供分子依据,并在培育新品种方面发挥重要的应用价值。
在20个世纪,以Anderson[11]为代表的科学家认为杂交物种的形态特征能够从父母本中遗传继承,从而可以通过形态特征实现对杂交物种的鉴定;随着研究的深入,人们发现当杂交个体缺乏中间形态或父母本形态特征时,将导致有效的形态特征过少[12],并且由于形态特征易受环境因素的影响,导致鉴定杂交物种的准确度很低,因此该方法逐步淡出了人们的视野。
鉴于形态学特征不能很好的鉴定杂交物种,人们开始寻找新的遗传标记,如利用次级代谢物进行杂交物种的鉴定提供了较为可靠的证据。比较成熟的标记物主要包括酚类、萜类、生物碱、异硫氰酸酯和黄酮类化合物,其中黄酮类化合物使用的频率最高[12]。1951年,Zobel[13]首次将次级代谢产物标记用于杂交物种的识别;1962年,Alston和Turner[14]的研究将这个方法有效推广。然而,人们意识到由于次级代谢产物产生的遗传机制非常复杂,所以无论是被用作定性指标还是定量指标,在鉴定杂交的准确性上均存在问题[15-16],限制了次级代谢物方法在杂交物种研究中的具体应用。此外,染色体数目同样被作为鉴定杂交的方法,该方法认为杂交个体总是伴随着染色体数目的加倍[17]。现实中也确实存在异源多倍化杂交的例子,如菊科的婆罗门参属[18]、禾本科的米草属[19]等。由于存在杂交个体与父母本具有相同染色体数目的可能性,单纯使用染色体数目方法会严重低估杂交的频率。
基于基因组分子标记的群体遗传学法和分子系统发育关系法,极大提高了杂交物种起源研究的分类深度和亲本定位的准确性。其中群体遗传学的方法主要包括NEWHYBRIDS[20]和INTROGRESS[21],在研究范围涵盖杂交物种父母本的情况下,该方法可以清楚的展示杂交物种保留父母本遗传信息的比例,但在鉴定杂交物种方面存在限制;系统发育关系法可以很好的解决此问题,目前使用系统发育关系法鉴定杂交的依据主要包括两点:一是杂交物种在不同单基因树中的位置差异[22];二是杂交物种在叶绿体系统发育关系与核基因的系统发育关系中的位置冲突[23]。前者在使用大量标记基因时,过多的单基因树拓扑结构差异会导致该方法的可操作性较差;后者虽然可以提供杂交物种的父母本信息[24],但由于叶绿体基因相对保守,物种间序列差异较小,导致叶绿体基因不能解决快速分化或分类阶元较低的类群的系统发育关系[25-26]。综上所述,目前仍旧缺少一种基于序列信息鉴定杂交的高效方法,因此我们尝试使用基于比较基因组学的方法来鉴定十字花科内部的杂交现象。
十字花科是公认的自然大科,属于世界广布植物。全科约有338个属,3 700多个种[25]。其中,我国分布约有102个属,421个种[27]。该科不仅包含许多观赏作物(如紫罗兰)和重要的经济作物(如油菜、芥菜等),同时还是分子生物学和进化发育生物学研究广泛使用的体系,包括了模式生物拟南芥等。因此十字花科的研究是广泛的、重要的。十字花科中杂交现象普遍存在[28],例如芸苔属中3个基本种(Brassica nigra,Brassica oleracea and Brassica rapa)两两杂交而产生的3个杂交种(Brassica carinata,Brassica napus and Brassica juncea)[29],同时存在一些属(Cardamine,Boechera)易频繁的发生杂交事件[30]。因此十字花科对于杂交现象的研究是非常好的材料。
十字花科的杂交事件一直是人们研究的热点。但是,目前仍缺少一种高效的鉴定杂交物种的方法,本研究选取28个十字花科物种及两个外类群物种作为研究材料,基于比较基因组学的方法来鉴定杂交物种,以期为深入研究十字花科杂交事件提供新的思路。
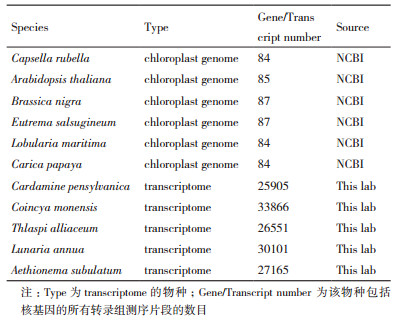
1 材料与方法 1.1 材料本研究使用了28个十字花科物种(涵盖16个族及两个未定族)和2个外类群物种。其中十字花科物种包括来自本实验室测序的22个转录组信息和来自公共数据库[31]的6个基因组信息;外类群物种包括来自本实验室测序的Cleome serrulata和来自公共数据库的Carica papaya(表 1)。

Huang等[32]利用56个物种的113个低拷贝核基因构建了稳定的十字花科系统发育关系,并将十字花科分为6个主要的支系(A-F支)。我们依据该结果确定了本研究所用的30个物种的系统发育关系(图 3)。其中,A支包含8个物种,B支包含10个物种,C支包含5个物种,D、E支各包括2个物种,F支包含1个物种以及两个外类群物种。

|
| 图 3 基于序列比对法获得拟南芥、诚实花best-hit分布图 (a)数字代表拟南芥(Arabidopsis thaliana)和诚实花(Lunaria annua)的best-hit数量在物种树上的映射情况;(b)拟南芥best-hit分布数量的汇总;(c)诚实花best-hit分布数量的汇总。 |
利用Trinity[33]对转录组数据进行拼接(使用默认参数),使用TGICL[34](参数:-p 0.98 -l 40 -v)进一步拼接以获得更长的unigene序列。接下来对30个物种的蛋白质序列进行两两的all-against-all Blastp比对分析,过滤掉相似度较低的比对结果。最后使用MCL[35]对比对结果进行聚类得到30个物种的同源基因簇(Orthogroup),分析流程见图 1-A。

|
| 图 1 核基因(A)与叶绿体基因(B)分析流程 A:基于核基因的序列比对法、单基因树映射物种树法分析流程;B:基于叶绿体基因重建系统发育关系法分析流程 |
同源基因簇的筛选标准:(1)序列的平均长度≥600 bp;(2)A、B、C三个类群的物种覆盖度分别达到80%(A、B、C类群在同源基因簇中分别应包含至少7、8、4个物种);(3)去除trimAl(参数:-automated1 -resoverlap 0.5 -seqoverlap 50)后序列过短的同源基因簇,最终得到7 179个同源基因簇。
使用RAxML[36]对上述同源基因簇构建单基因树(参数:-m GTRCAT -f a -x 1 -N 100),并对十字花科物种树进行映射,映射方法见图 2,仅保留bootstrap≥70的姐妹类群,最终得到各物种的姐妹类群的分布情况,筛选杂交物种,用于后续分析。

|
| 图 2 单基因树映射物种树模型及基于该方法获得拟南芥、诚实花的分布图 (a)(b)(c)(d)为单基因树映射物种树模型。其中,(a)为物种树。(b)(c)为两个单基因树。(d)为单基因树映射物种树的转换表,其中Cluster为单基因树编号;Sister1与Sister2是单基因树中互为姐妹类群的物种或者节点;BS为单基因树中姐妹类群的支持度(bootstrap),仅保留BS ≥ 70;Level1与Level2是Sister1与Sister2在物种树中的映射(即物种树中的Level1节点能够包含单基因树中Sister1节点范围内的所有物种且Level1自身包含最少的物种。以d中的第二行为例,Sister1为N0,Level1为P1,N0包含A1和C1两个物种的基因,那么物种树中的Level1应包含A和C物种,这样的节点有P1、P2、P4、P5,同时,要求Level1中包含最少的物种,所以,Level1为P1)。假定查看物种E的姐妹类群分布情况,则P0和P1两个节点各计分一次。(e)(f)分别为拟南芥与诚实花单基因映射物种树分布图(数字代表诚实花的姐妹类群数量的前10名) |
将同源基因簇中特定物种的蛋白质序列与其他物种的所有序列进行Blastp(使用默认参数)搜索,仅保留每条比的最匹配序列(best-hit),接下来将7 179个同源基因簇重复此操作,并汇总所有保留的比对结果,然后按照query的物种名进行分类,统计各物种比对的best-hit物种名的数量情况,最终将各物种的best-hit的分布数量映射到物种树上,得到best-hit分布图。
1.2.4 叶绿体/核基因系统发育关系对比法叶绿体基因具有序列保守、单亲遗传等特点[37],是植物系统发育关系研究的常用材料[38]。本方法使用10个十字花科物种和一个外类群物种,其中十字花科物种包括5个叶绿体基因组信息和5个转录组信息,外类群Carica papaya为叶绿体基因组信息(表 2)。
我们以拟南芥叶绿体基因(共85个叶绿体基因,其中反向重复序列为7对,长单拷贝与短单拷贝的序列为71个)中7个重复序列和71个非重复序列共计78个基因作为叶绿体基因库,对11个物种的所有序列进行Tblastn序列比对,过滤掉非叶绿体基因(相邻得分差大于最高得分的10%,则认为后续序列为非叶绿体基因);筛选包含杂交物种的同源基因簇,并将每个物种的基因顺序串联,构建super matrix;使用RAxML构建系统树(参数:-m GTRCAT -f a -x 1 -N 100),分析流程见图 1-B,最后将叶绿体基因树与核基因树进行比较,寻找冲突位点,确定可能的杂交物种及可能的父母本来源。
1.2.5 功能富集分析对于杂交物种,我们对其父源、母源基因进行功能富集的比较分析。利用MapMan[39]注释体系获取拟南芥的注释信息(http://mapman.gabipd.org/)。由于同一个同源基因簇中的基因在功能上高度相似,因此我们选取同源基因簇中的拟南芥基因代替杂交物种基因进行功能富集分析来表明该同源基因簇的功能,最后使用Excel对注释结果进行可视化处理。
2 结果 2.1 转录组的拼接与同源基因簇的筛选本研究共使用30个物种,其中23个物种为转录组数据,首先对转录组进行拼接,统计转录本数目并汇总所有物种的基因数目,发现30个物种除Brassica rapa和Parolinia intermedia外,物种包含基因/转录本数在24 000-35 000之间(表 1)。
对30个物种的890 487条基因序列进行all-against-all Blastp序列比对,过滤掉query与hit长度存在显著差异的序列,并将比对结果通过MCL进行聚类分析,最终得到覆盖30个物种的30 091个同源基因簇。每个同源基因簇平均包括15个物种(物种覆盖度为50%)、26个基因(序列平均长度为853 bp)。
为了获得更加稳定的单基因树,对同源基因簇序列平均长度和物种覆盖度进行过滤,当序列的平均长度≥600 bp,可以筛选到15 624个同源基因簇,同源基因簇保留率为52%。进一步对物种覆盖度进行过滤,筛选得到7 210个同源基因簇,同源基因簇保留率为24%。最后去除trimAl后序列过短的同源基因簇得到7 179个同源基因簇,平均每个同源基因簇包括29个物种(物种覆盖度为97%)、59个基因(序列平均长度为1 217 bp)。使用RAxML对同源基因簇构建单基因树,得到7 179个单基因树。
2.2 单基因树映射物种树法将7 179个单基因树映射到十字花科物种树,从基因树中反映出的物种关系(即给定物种的姐妹类群分布,附图 1-6)出发,对单基因的物种来源做统计。结果可以分为两类:第一类以模式生物拟南芥(Arabidopsis thaliana)为代表,其姐妹类群分布相对集中;第二类以诚实花(Lunaria annua)为代表,其姐妹类群分布相对分散,在十字花科基部和内部的多个分支上均有所分布。以拟南芥和诚实花为例(图 2),在7 179个单基因树中,拟南芥单基因的姐妹类群在物种树上共计映射5 639次,且主要分布在A类群a亚分支,其包含3个物种,2个节点,覆盖拟南芥姐妹类群分布数量的前五名,占映射总数的89.4%,且与第六名以及之后的占比差异较大;而诚实花的姐妹类群在物种树上共计映射3 693次,其姐妹类群数量前五名只占总数量的63%,前十名占比为81.7%,各种类占比呈现平稳下降,与拟南芥形成鲜明对比(表 3)。
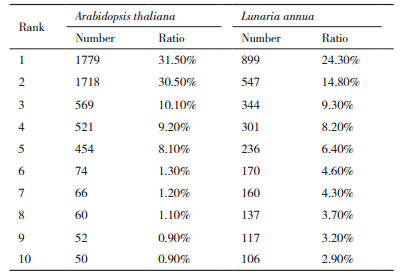
总体来说,诚实花的姐妹类群的分布较分散,除集中分布在C类群外,还在A类群、B类群的祖先处以及十字花科基部类群均有所分布。该结果表明诚实花与其他类群可能存在一定的基因交流,为揭示十字花科内部的杂交物种提供了线索。
2.3 序列比对法(best-hit法)针对杂交物种诚实花,我们使用序列比对法对其进行分析,得到其best-hit的分布图,并与拟南芥进行对比(图 3)。在7 179个同源基因簇中,共包含18 723个拟南芥基因,其中6个主要支系的26个物种包含18 394个best-hits,平均每个物种best-hit的基因数为707,其中在A支系平均每个物种的best-hits基因数为2 315,远远高于平均值以及B、C类群的best-hit基因数。对于诚实花,7 179个同源基因簇中共包含15 727个基因,其中6个主要支系的27个物种包含15 698个best-hits,平均每个物种best-hits的基因数为581,诚实花所在C类群best-hit基因数为519,与平均值以及A、B类群相比不存在显著性差异(P值分别为0.41、0.23和0.35)。通过best-hit法,我们发现诚实花的best-hit分布比拟南芥等系统位置明确的物种更加分散,间接支持诚实花可能是一个杂交物种。
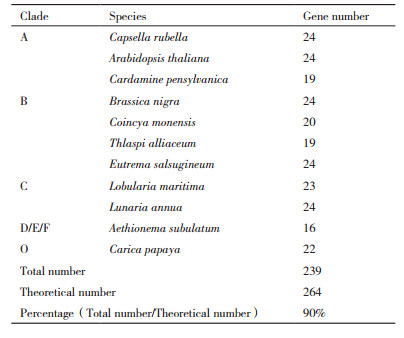
2.4 叶绿体/核基因系统发育关系对比法以拟南芥78个叶绿体基因作为基因库,对11个物种的叶绿体基因组/转录组信息进行序列比对,过滤掉非叶绿体基因,筛选含有杂交物种的同源基因簇,最终得到11个物种的24个叶绿体基因,其中Aethionema subulatum包含最少叶绿体基因数为16个。11个物种共包含239个叶绿体基因,物种的基因覆盖度达90%(表 4)。将每个物种的基因顺序串联,构建super matrix,最终使用RAxML构建十字花科叶绿体的系统发育关系(图 4)。通过与十字花科核基因系统发育关系进行对比,我们发现诚实花在核基因系统发育关系中的姐妹类群为C类群,在叶绿体系统发育关系中,与B、C结合类群互为姐妹类群。因此,我们认为诚实花可能是一个杂交物种,其母本可能来自于B、C结合类群的共同祖先,父本可能来自于C类群。

|
| 图 4 十字花科核基因树、叶绿体基因树以及单基因树个例展示 (a)使用 super matrix 的方式构建十字花科核基因树 ;(b)使用 super matrix 的方式构建十字花科叶绿体基因树 ;(c)和(d)分别为 1号、10号节点核基因树。 |
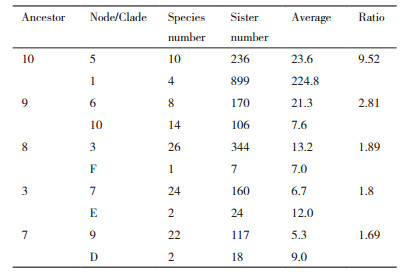
在7 179个同源基因簇中,有4 234个同源基因簇包含合格的诚实花基因(BS≥70的姐妹类群中的诚实花基因)。由于同一个同源基因簇中的基因在功能上高度相似,因此使用拟南芥基因代替诚实花进行功能富集分析。在4 234个同源基因簇中共包含5 292个拟南芥基因,其中前10名(图 2-f)包含4 105个基因,占比为77.6%。其中39.1%的基因与父本来源的C类群基因关系最近;38.5%的基因与C类群以外类群的基因最近,特别是来自B、C结合类群共同祖先的10号节点(占比3%)支持其作为诚实花的母本来源。结合诚实花单基因树映射物种树的姐妹类群分布情况,我们将图 2-f中1、2、4号节点定义为杂交父本,将3、7、8、9、10号节点定义为杂交母本,然后对杂交物种诚实花的父母本进行功能富集分析(图 5),最后我们发现父本主要富集在生物降解、次级代谢产物、RNA调控等途径上,表明其可能通过RNA调控生物降解、次级代谢过程以适应胁迫环境;母本主要富集在代谢途径上,尤其是碳水化合物、核酸代谢,表明其可能通过增强自身代谢获取相对竞争优势。

|
| 图 5 诚实花基因的父源、母源基因功能富集的比较分析 富集分析检验的P值已进行-log10(P)转换,图中黑色虚线对应P值为0.01 |
进化研究表明,由于受到基因突变速率差异、横向基因转移以及重复基因丢失拷贝等因素的影响,单基因树的拓扑结构可能与物种关系存在一定程度的差异。以相对准确的物种进化历史作为参照,从单基因树中包含的重要进化信息[40-41]出发,寻找可能的序列特征以追溯历史上发生过的杂交等复杂事件是本研究的主要目的。随着二代测序的发展,基因组信息得以迅速积累,促进了构建物种关系和基因进化历史等相关研究的发展[42]。分子进化信息位点数量的快速增长,将有助于发现不同物种进化历史上的杂交事件和其它种间基因交流方式。
在本研究中,我们基于筛选后的7 179个十字花科同源基因簇数据集,结合单基因树映射物种树分析方法,对所有的单基因树进行整合,绘制出各物种的姐妹类群分布图用于鉴定十字花科内部的杂交事件。由于该方法主要依据单基因树的拓扑结构信息,因此单基因树的建树质量对于结果的准确性有显著影响。为此在对同源基因簇进行过滤时,我们使用更严格的条件(序列长度≥600 bp,A、B、C各分支物种覆盖度≥80%),最终同源基因簇的保留率为23.8%,对重要节点(父母本来源的节点)同源基因簇所构建的单基因树进行检查(图 4-c、d,附图 7),我们发现使用该方法过滤后的同源基因簇所构建的单基因树与十字花科物种树拓扑结构基本一致,且支持度较高,能够满足该方法的使用。
在单基因树映射物种树的方法中,诚实花的姐妹类群主要分布在父本来源的C类群(1、2、4号),共计映射1 790次,占比39.1%,而母本来源的10号节点共映射106次,占比只有3%。我们认为造成杂交物种母本来源的基因较少的原因主要由于父母本发生杂交事件,母本进化速率加快,从而导致姐妹类群的分布分散到十字花科的基部类群(3、7、8、9号节点),因此在对父母本进行功能富集分析时,将3、7、8、9、10五个节点的信息归于母本来源。
前人研究表明,单基因树与物种树存在差异受水平基因转移、谱系分选和杂交等多种因素影响[43, 8]。其中,水平基因转移是相对于亲代到子代的垂直遗传方式而言,指不通过生殖进行的、跨物种的遗传信息交流的方式[44]。在植物核基因之间水平基因转移发生的频率低[45],因此对姐妹类群分布情况不会造成显著影响;谱系分选指由于物种分化时间短,祖先基因的多态性在分化物种中随机固定下来的过程。由于谱系分选对单基因树拓扑结构的影响与物种分化后发生杂交事件的影响是一致的,所以对于杂交物种的鉴定,谱系分选的影响不容忽略。
考虑到谱系分选在分化物种中随机固定的特性,Huson等[46]基于单基因树拓扑结构进行统计检测,假定{{a,b},c}是一个物种树,如果单基因树冲突是由于谱系分选造成的,那么{{a,c},b}{{b,c},a}两种拓扑结构应该具有相同的出现频率。鉴于此,我们分别对图 2-f中的3、7、8、9、10五个节点分化后诚实花的姐妹类群在各节点分支处的数量进行统计分析,具体信息见表 6。其中10号节点分化后形成1、5号节点,1号节点包括10个物种,有236个诚实花的姐妹类群覆盖在该节点,平均每个物种包含23.6个姐妹类群;5号节点包括4个物种,有899个诚实花姐妹类群覆盖,平均每个物种包含224.8个姐妹类群,两分支节点平均值相差9.52倍,显然不符合谱系分选的比例要求。因此,我们认为C类群是诚实花杂交事件的一个亲本。9号节点分化后形成6、10号节点,6号节点每个物种平均包含21.3个姐妹类群,10号节点每个物种平均包含7.6个物种,两分支节点平均值相差2.8倍,不符合谱系分选的比例要求。因此,我们认为B、C结合类群的共同祖先是诚实花杂交事件的另一个亲本。3、7、8号节点由于物种分化后的E、D、F分支只有1到2个物种,随机因素对其结果影响较大,倍数差异可信度较差,未来可能需要提供更多的物种或者通过其他证据进行验证。
综上所述,我们认为诚实花杂交的父本来自C类群,母本来自B、C结合类群的共同祖先,而诚实花姐妹类群较多的分布在3、7、8号节是谱系分选的影响还是杂交的影响,有待寻找更多的证据来进行验证。
4 结论本研究通过选取28个十字花科物种(涵盖16个族及两个未定族)及2个外类群物种作为研究材料,采用比较基因组学和系统发育基因组学的研究手段,在叶绿体和核基因两个方向上联合分子表型信息鉴定十字花科内的杂交事件。
主要结论包括:(1)依据单基因树映射物种树的结果,将十字花科物种分为两类,其一以拟南芥为代表,其姐妹类群分布相对集中;其二以诚实花为代表,其姐妹类群分布分散,暗示该物种基因可能存在多种来源。
(2)结合最佳比对法和叶绿体/核基因系统发育关系对比法,我们认为诚实花可能是一个杂交物种,其39.1%的基因与父本来源的C类群基因关系最近;38.5%的基因与C类群以外类群的基因最近,特别是来自B、C结合类群共同祖先的10号节点(占比3%)支持其作为诚实花的母本来源。
(3)对杂交物种诚实花的父母本进行功能富集分析,最后我们发现父本主要富集在生物降解、次级代谢产物、RNA调控等途径上,表明其可能通过RNA调控生物降解、次级代谢过程以适应胁迫环境;母本主要富集在代谢途径上,尤其是碳水化合物、核酸代谢等方面,表明其可能通过增强自身代谢获取相对竞争优势。
注:论文附图 1-7见电子版(http://biotech.caas.cn)
| [1] |
Mallet J. Hybridization as an invasion of the genome[J]. Trends Ecol Evol, 2005, 20(5): 229-237. DOI:10.1016/j.tree.2005.02.010 |
| [2] |
Arnold ML. Evolution Through Genetic Exchange[M]. Oxford University Press, 2006.
|
| [3] |
Mooney HA, Cleland EE. The evolutionary impact of invasive species[J]. Proc Natl Acad Sci USA, 2001, 98(10): 5446-5451. DOI:10.1073/pnas.091093398 |
| [4] |
Schierenbeck KA, Ellstrand NC. Hybridization and the evolution of invasiveness in plants and other organisms[J]. Biol Invasions, 2009, 11(5): 1093. DOI:10.1007/s10530-008-9388-x |
| [5] |
Levin DA, Francisco-Ortega J, Jansen RK. Hybridization and the extinction of rare plant species[J]. Conserv Biol, 1996, 10(1): 10-16. DOI:10.1046/j.1523-1739.1996.10010010.x |
| [6] |
Rhymer JM, Simberloff D. Extinction by hybridization and introgression[J]. Annu Rev Ecol Evol Syst, 1996, 27(1): 83-109. DOI:10.1146/annurev.ecolsys.27.1.83 |
| [7] |
Lopez-Caamal A, Tovar-Sánchez E. Genetic, morphological, and chemical patterns of plant hybridization[J]. Rev Chil Hist Nat, 2014, 87(1): 1-15. |
| [8] |
Som A. Causes, consequences and solutions of phylogenetic incongruence[J]. Brief Bioinform, 2014, 16(3): 536-548. |
| [9] |
Fu DH, Xiao ML, Hayward A, et al. Utilization of crop heterosis:A review[J]. Euphytica, 2014, 197(2): 161-173. DOI:10.1007/s10681-014-1103-7 |
| [10] |
Song Y, Zhang Z, Tan XJ, et al. Association of the molecular regulation of ear leaf senescence/stress response and photosynthesis/metabolism with heterosis at the reproductive stage in maize[J]. Sci Rep, 2016, 6: 1-12. DOI:10.1038/s41598-016-0001-8 |
| [11] |
Anderson E. Introgressive hybridization[J]. Biol Rev, 1953, 28(3): 280-307. |
| [12] |
Rieseberg LH, Ellstrand NC. What can molecular and morphological markers tell us about plant hybridization?[J]. Crit Rev Plant Sci, 1993, 12(3): 213-241. |
| [13] |
Zobel B. Oleoresin composition as a determinant of pine hybridity[J]. Bot Mag, 1951, 113(2): 221-227. |
| [14] |
Alston RE, Turner BL. New techniques in analysis of complex natural hybridization[J]. Proc Natl Acad Sci USA, 1962, 48(2): 130-137. DOI:10.1073/pnas.48.2.130 |
| [15] |
Cheng DD, Vrieling K, Klinkhamer PGL. The effect of hybridization on secondary metabolites and herbivore resistance:Implications for the evolution of chemical diversity in plants[J]. Phytochem Rev, 2011, 10(1): 107-117. DOI:10.1007/s11101-010-9194-9 |
| [16] |
Orians CM. The effects of hybridization in plants on secondary chemistry:Implications for the ecology and evolution of plant - Herbivore interactions[J]. Am J Bot, 2000, 87(12): 1749-1756. DOI:10.2307/2656824 |
| [17] |
Harlan JR, Dewet JM, On Ö. Winge and a prayer:the origins of polyploidy[J]. Bot Rev, 1975, 41(4): 361-390. DOI:10.1007/BF02860830 |
| [18] |
Roose ML, Gottlieb LD. Genetic and biochemical consequences of polyploidy in Tragopogon[J]. Evolution, 2017, 30(4): 818-830. |
| [19] |
Strong DR, Ayres DR. Ecological and evolutionary misadventures of Spartina[J]. Annu Rev Ecol Evol Syst, 2013, 44: 389-410. DOI:10.1146/annurev-ecolsys-110512-135803 |
| [20] |
Anderson EC, Thompson EA. A model-based method for identifying species hybrids using multilocus genetic data[J]. Genetics, 2002, 160(3): 1217-1229. |
| [21] |
Gompert Z, Alex Buerkle C. Introgress:A software package for mapping components of isolation in hybrids[J]. Mol Ecol Resour, 2010, 10(2): 378-384. DOI:10.1111/men.2010.10.issue-2 |
| [22] |
Linder CR, Rieseberg LH. Reconstructing patterns of reticulate evolution in plants[J]. Am J Bot, 2004, 91(10): 1700-1708. DOI:10.3732/ajb.91.10.1700 |
| [23] |
Guo YL, Ge S. Molecular phylogeny of Oryzeae(Poaceae)based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes[J]. Am J Bot, 2005, 92(9): 1548-1558. DOI:10.3732/ajb.92.9.1548 |
| [24] |
Liu M. Phylogenetic reconstruction of tribal relationships in Asteroideae(Asteraceae)with low-copy nuclear genes[J]. Chinese Bull Bot, 2015, 50: 549-564. |
| [25] |
Jansen RK, Cai Z, Raubeson LA, et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns[J]. Proc Natl Acad Sci USA, 2007, 104(49): 19369-19374. DOI:10.1073/pnas.0709121104 |
| [26] |
Moore MJ, Soltis PS, Bell CD, et al. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots[J]. Proc Natl Acad Sci USA, 2010, 107(10): 4623-4628. DOI:10.1073/pnas.0907801107 |
| [27] |
Warwick SI, Al-Shehbaz IA, Sauder CA. Phylogenetic position of Arabis arenicola and generic limits of Aphragmus and Eutrema(Brassicaceae)based on sequences of nuclear ribosomal DNA[J]. Can J Bot, 2006, 84(2): 269-281. DOI:10.1139/b05-161 |
| [28] |
Stace CA. Hybridization and the Flora of the British Isles[M]. Academic Press, 1975.
|
| [29] |
Nagaharu U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization[J]. Jpn J Bot, 1935, 7(7): 389-452. |
| [30] |
Marhold K, Lihová J. Polyploidy, hybridization and reticulate evolution:lessons from the Brassicaceae[J]. Plant Syst Evol, 2006, 259(2-4): 143-174. DOI:10.1007/s00606-006-0417-x |
| [31] |
Goodstein DM, Shu SQ, Howson R, et al. Phytozome:A comparative platform for green plant genomics[J]. Nucleic Acids Res, 2012, 40(D1): 1178-1186. DOI:10.1093/nar/gkr944 |
| [32] |
Huang CH, Sun RR, Hu Y, et al. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution[J]. Mol Biol Evol, 2016, 33(2): 394-412. DOI:10.1093/molbev/msv226 |
| [33] |
Grabherr MG, Haasl BH, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nat Biotechnol, 2011, 29(7): 644-652. DOI:10.1038/nbt.1883 |
| [34] |
Pertea G, Huang XQ, Liang F, et al. TIGR gene indices clustering tools(TGICL):A software system for fast clustering of large EST datasets[J]. Bioinformatics, 2003, 19(5): 651-652. DOI:10.1093/bioinformatics/btg034 |
| [35] |
Enright AJ, Dongen SV, Ouzounis CA. An efficient algorithm for large-scale detection of protein families[J]. Nucleic Acids Res, 2002, 30(7): 1575-1584. DOI:10.1093/nar/30.7.1575 |
| [36] |
Stamatakis A. RAxML-VI-HPC:Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models[J]. Bioinformatics, 2006, 22(21): 2688-2690. DOI:10.1093/bioinformatics/btl446 |
| [37] |
Richard G, Jeffrey D. Chloroplast DNA systematics:A review of methods and data analysis[J]. Am J Bot, 2009, 81(9): 1205-1224. |
| [38] |
Moore MJ, Soltisb PS, Bell CD, et al. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots[J]. Proc Natl Acad Sci USA, 2010, 107(10): 4623-4628. DOI:10.1073/pnas.0907801107 |
| [39] |
Thimm O, Bläsing O, Gibon Y, et al. Mapman:a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes[J]. Plant J, 2004, 37(6): 914-939. DOI:10.1111/tpj.2004.37.issue-6 |
| [40] |
de Queiroz A, Donoghue MJ, Kim J. Separate versus combined analysis of phylogenetic evidence[J]. Annu Rev Ecol Syst, 1995, 26(1): 657-681. DOI:10.1146/annurev.es.26.110195.003301 |
| [41] |
Wendel J, Doyle J. Phylogenetic Incongruence: Window into Genome History and Molecular Evolution[M]. //Soltis DE, Soltis PS, Doyle JJ(eds)Molecular Systematics of Plants Ⅱ. Springer, 1998.
|
| [42] |
Rokas A, Carroll SB. Bushes in the tree of life[J]. PLoS Biol, 2006, 4(11): 1899-1904. |
| [43] |
Jeffroy O, Brinkmann H, Delsuc F, et al. Phylogenomics:the beginning of incongruence?[J]. Trends Genet, 2006, 22(4): 225-231. DOI:10.1016/j.tig.2006.02.003 |
| [44] |
Gao C, Ren XD, Mason AS, et al. Horizontal gene transfer in plants[J]. Funct Integr Genomics, 2014, 14(1): 23-29. DOI:10.1007/s10142-013-0345-0 |
| [45] |
石磊, 张明慧, 陈虞超, 等. 植物中的水平基因转移[J]. 植物学报, 2016, 51(4): 542-559. DOI:10.11983/CBB15121 |
| [46] |
Huson DH, Klöpper T, Lockhart PJ, et al. Reconstruction of reticulate networks from gene trees[M]. Berlin Heidelberg: Springer, 2005.
|