




胚胎干细胞(Embryonic stem cell,ESC)是在胚胎发育早期动物囊胚中存在的一群尚未分化的原始细胞[1],具有发育上的全能性,并能在体外无限增殖。胚胎干细胞的干性维持和谱系分化受到多层次、网络化的调控,包括关键转录因子[2]、信号通路[3-4]和表观遗传学修饰[5]等。有报道指出,miRNA这类小分子RNA在胚胎发育、干细胞分化、细胞凋亡及肿瘤发生发展等多种生物学进程中都起着重要的作用[6]。miR-22是由22个核苷酸组成的非编码RNA,其在多种物种中序列高度保守。据报道,miR-22在不同类型的细胞中具有不同的生物学功能,在肿瘤细胞中,miR-22作为肿瘤抑制因子[7-9],抑制细胞增殖、迁移[10];在心肌细胞中,miR-22高表达促进自噬、抑制细胞凋亡,从而保护心肌细胞免受饥饿损伤[11];miR-22通过抑制BMP7促进肝硬化的发展[12];在小鼠胚胎干细胞中,miR-22抑制MECP2表达,从而促进胚胎干细胞向平滑肌方向分化[13]。然而,miR-22对小鼠胚胎干细胞干性维持、自我更新和谱系分化的系统研究相对较少,有待进一步深入探索。
CRISPR/Cas9技术源于嗜热型古细菌为抵御外源DNA、病毒入侵所进化出的免疫系统中的规律成簇的间隔短回文重复序列[14-15],这种短回文重复序列与许多外源病毒或DNA的序列互补。CRISPR/Cas9系统中,sgRNA通过自身NGG前20个碱基与基因组中的靶位点碱基互补配对结合,将Cas9蛋白导向靶位点,发挥其核酸内切酶作用,将DNA双链切断[16]。断裂的双链DNA可以进行两种修复,非同源性末端接合(Non-homologous end joining,NHEJ)和同源介导修复(Homology Directed Repair,HDR)。利用CRISPR/Cas9技术进行靶基因的敲除,具有适用面积广,敲除效率高,操作简单和节省时间等优势,已经在不同物种中得到广泛应用[17-22]。
为研究miR-22在小鼠胚胎干细胞中的功能,我们拟利用CRISPR/Cas9技术获得miR-22敲除的小鼠胚胎干细胞。首先,利用点突变和搭桥PCR等方法构建neomycin(neor)抗性的sgRNA表达载体(PGL3-2sgRNA-neor),并设计靶向miR-22的sgRNA,克隆至该载体中;其次,将靶向敲除miR-22的sgRNA载体电转至稳转Cas9的细胞系中,并通过药物筛选、PCR基因型鉴定和RT-qPCR等方法筛选miR-22敲除的胚胎干细胞;最后,对miR-22敲除克隆进行功能分析。该项工作首次利用CRISPR/Cas9技术构建miR-22缺失的小鼠胚胎干细胞,对于丰富miRNA在胚胎干细胞中的功能研究有着重要意义。
1 材料与方法 1.1 材料 1.1.1 仪器Veriti 96热循环仪、HE-120多功能水平电泳槽、Tanon2500全自动数码凝胶图像分析系统、5427R离心机、NanoDrop 2000超微量生物检测仪、SW-CJ-1FD洁净工作台、DHP-9162恒温培养箱、THZ-D恒温摇床、BSC-1300IIA2生物安全柜、3111二氧化碳培养箱、Nucleofector 2b细胞核转染系统、ViiA7实时荧光定量PCR仪、OLYMPUS IX83荧光倒置显微镜。
1.1.2 实验材料E14小鼠胚胎干细胞细胞系(简称E14 mES)和PL451载体(英国剑桥大学Sanger研究所惠赠),pGL3-U6-2sgRNA-ccdB-EF1a-puromycin sgRNA载体(简称pGL3-2sgRNA-puro,苏州大学徐璎老师实验室惠赠),pUC57载体,限制性内切酶Hind Ⅲ、Xba Ⅰ和Bsm BI购自美国NEB公司,T4连接酶购自美国Promega生物技术有限公司,TransStart FastPfu DNA Polymerase购自全式金生物技术有限公司,KnockOut DMEM购自Thermo Fisher Scientific公司,血清购自美国LabTech公司,ESGRO mLIF、Neomycin(G418,新霉素)和胰蛋白酶购自Sigma公司。
1.2 方法 1.2.1 G418抗性sgRNA表达载体的构建稳转Cas9的胚胎干细胞为puro抗性,为提高筛选效率拟构建以neor为抗性筛选标记的sgRNA表达载体。策略为以PL451载体为模板,将PGK-neor抗性基因克隆至pGL3-2sgRNA-puro载体中,从而将sgRNA载体原有的puro筛选标记替换为neor筛选标记。此外,靶基因sgRNA载体的构建需用Bsm BI限制性内切酶,而PGK-neor启动子区域含有一个Bsm BI的识别位点。为解决这一问题,我们采用搭桥PCR的方法,设计搭桥引物,实现PGK-neor中Bsm BI酶切位点识别序列由“CGTCTC”突变为“CGTATC”。搭桥PCR所用引物,见表 1。

具体载体构建步骤如下:以PGK-neor-F1和R1,PGK-neor-F2和R2为引物,以PL451载体为模板,分别扩增大小为429 bp和1 161 bp的PCR产物,依次命名为片段Ⅰ和Ⅱ(图 1-A)。以PGK-neor-F1和PGK-neor-R2为引物,以片段Ⅰ和Ⅱ为模板(两个片段的摩尔比1:1),扩增得到Bsm BI酶切位点识别序列突变的PGK-neor序列Ⅲ(图 1-A)。随后,将序列Ⅲ经Hind Ⅲ和Xba Ⅰ双酶切,克隆至Hind Ⅲ/Xba Ⅰ酶切后的pGL3-2sgRNA-puro载体中,得到neor筛选标记的sgRNA载体,命名为pGL3-2sgRNA-neor。

|
| 图 1 pGL3-2sgRNA-neor载体的构建 A:点突变和搭桥PCR模式图(用引物F1和R1,F2和R2,F1和R2进行PCR扩增,得到产物分别为Ⅰ,Ⅱ和Ⅲ;经过搭桥PCR,将Bsm BI识别位点“CGTCTC”突变成“CGTATC”);B:点突变、搭桥PCR凝胶电泳图(Ⅰ、Ⅱ和Ⅲ片段大小分别为429 bp、1 161 bp和1 565 bp);C:Bsm BI识别序列点突变测序结果显示,碱基由“C”突变成“A” |
在miR-22敲除区域的上下游分别设计2个sgRNA,sgRNA靶位点是符合(N)20NGG的序列,sgRNA通过http://crispr.mit.edu:8079/网站设计。引物的5' 端引入保护碱基、Bsm BI的酶切位点,3' 端引入U6启动子序列引物,引物序列见表 2。

miR-22靶向敲除的sgRNA载体构建步骤如下:以miR-22 sgRNA-F1和R1,miR-22 sgRNA-F2和R2为引物,以pUC57载体为模板进行PCR,分别获得sgRNA-1-U6-sgRNA-3和sgRNA-2-U6-sgRNA-4的串联片段。将串联片段通过Bsm BI酶切分别克隆至pGL3-2sgRNA-neor载体,从而得到pGL3-sgRNA-1-U6-sgRNA-3-neor和pGL3-sgRNA -2-U6-sgRNA-4-neor两种载体。
1.2.3 小鼠胚胎干细胞的培养小鼠胚胎干细胞用ES培养基[DMEM中加入10% fetal bovine serum,2 mmol/L L-glutamine(Gbico),50 mg/mL penicillin,80 mg/mL streptomycin,0.1 mmol/L 2-mercaptoethanol(Sigma),LIF(Millipore,ESG1107)]培养,于37℃,5% CO2条件下培养。
1.2.4 电转及G418筛选电转前1 d,将E14 mES细胞按50%的密度接种至六孔板中。次日,细胞密度达到70%-80%时进行电转。电转48 h后用终浓度为250 μg/mL的G418筛选,每隔2 d换一次液,筛选8 d左右挑取单克隆至96孔板中培养,待96孔板细胞密度达到80%,将96孔板细胞一分为二,一板用于提取DNA进行基因型鉴定,另一板细胞于液氮中冻存。
1.2.5 miR-22敲除基因型鉴定基因组DNA提取时,于96孔板中每孔加入50 μL裂解液(10 mmol/L Tris-HCl[pH 8.0],0.5 mmol/L EDTA,0.5% Triton X-100,0.5 mg/mL Proteinase K),65℃裂解3 h。加入150 μL沉降液(无水乙醇加3% 5 mol/L NaCl)沉降30 min,4 000 r/min离心15 min,弃上清。后经70%乙醇洗涤,离心,晾干后,加50 μL ddH2O溶解。基因型鉴定首先用敲除区域外部引物miR-22-F1和R1进行PCR,之后用敲除区域内部引物miR-22-F2和R2进一步验证。基因型鉴定所用引物序列见表 3。

RNA提取采用TRIzol法(TaRaKa,9109)。检测miR-22的表达,用TransScript® miRNA Frist-Strand cDNA Synthesis Super-Mix(Transgene,AT351)试剂盒进行逆转录,后用SYBR Premix Ex Taq™ Ⅱ(TaKaRa,RR820A)进行qPCR。检测干性相关因子Oct4、Sox2和Nanog的表达,用HiScript®Ⅱ Q RT SuperMix for qPCR(Vazyme,R223-01)进行逆转录反应,后用SYBR Premix Ex Taq™ Ⅱ进行qPCR。qPCR所用引物见表 4。

为构建neor筛选标记的sgRNA载体,并将PGK-neor启动子区域的Bsm BI识别位点破坏,我们采用点突变和搭桥PCR的方法:以PL451载体为模板,以F1,R1和F2,R2为引物进行PCR扩增,分别得到大小为429 bp的片段Ⅰ和1161 bp的片段Ⅱ(图 1-A,1-B)。以F1和R2为引物,以片段Ⅰ和Ⅱ为模板进行搭桥PCR,最终获得大小为1 565 bp的目的片段Ⅲ(图 1-A,1-B)。将片段Ⅲ和pGL3-2sgRNA-puro载体分别经Hind Ⅲ与Xba Ⅰ双酶切,后经连接、转化等步骤获得pGL3-2sgRNA-neor载体。测序结果证实pGL3-2sgRNA-neor载体中PGK-neor启动子区域的Bsm BI识别位点“CGTCTC”突变为“CGTATC”,说明Bsm BI酶切位点被破坏(图 1-C)。
2.2 miR-22靶向敲除所用sgRNA载体的构建为在小鼠胚胎干细胞中靶向敲除miR-22,在miR-22的上下游两侧分别设计两个sgRNA,命名为sgRNA-1、sgRNA-2(上游),sgRNA-3、sgRNA-4(下游)(图 2-A)。以miR-22 sgRNA-F1和R1,miR-22 sgRNA-F2和R2为引物,以pUC57为模板,进行PCR扩增,得到sgRNA-1-U6-sgRNA-3和sgRNA-2-U6-sgRNA-4的扩增产物。将上述片段经Bsm BI酶切后分别克隆至Bsm BI处理后的pGL3-2sgRNA-neor载体中。测序结果证实pGL3-sgRNA-1-U6-sgRNA-3-neor和pGL3-sgRNA-2-U6-sgRNA-4-neor载体构建成功(图 2-B)。

|
| 图 2 靶向敲除miR-22所用sgRNA载体的构建 A:靶向敲除miR-22所用sgRNA的位置分布(sgRNA-1、2位于miR-22敲除区域的上游,sgRNA-3、4位于下游);B:miR-22敲除所用sgRNA载体的模式图(sgRNA-1和3,sgRNA-2和4分别构建到两个sgRNA载体中;sgRNA表达载体的测序结果,黄色阴影区域为sgRNA序列) |
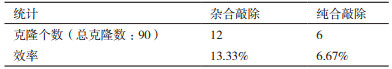
将靶向miR-22的sgRNA表达载体,电转至稳转Cas9的小鼠胚胎干细胞中,经neor筛选后,挑取单克隆并进行基因型鉴定。首先,用敲除区域外部引物miR-22-F1和R1进行PCR,发现克隆4、克隆19的PCR产物约为800 bp(图 3-A,3-B),并没有检测到1 216 bp的野生型条带,暗示miR-22可能被敲除;其次,用内部引物miR-22-F2和R2进一步验证,发现克隆4、克隆19均未得到304 bp的扩增产物,证实克隆4、克隆19为miR-22纯合敲除的胚胎干细胞克隆(图 3-A,3-C)。此外,对两个敲除克隆用引物F1和R1进行PCR扩增,产物构建TA克隆测序,发现克隆4和克隆19分别被敲掉529 bp和378 bp(图 4)。本实验共挑取了90个克隆,其中miR-22杂合敲除克隆12个、纯合敲除克隆6个,杂合敲除和纯合敲除的效率分别为13.33%和6.67%(表 5)。

|
| 图 3 miR-22敲除克隆的筛选及基因型鉴定 A:鉴定miR-22敲除所用引物的位置,F1和R1位于敲除区域外部,F2和R2位于敲除区域的内部;B:用外部引物F1和R1进行基因型鉴定,发现克隆4和克隆19的PCR产物明显变小;C:用内部引物F2和R2进行基因型鉴定,克隆4、19均未扩增出条带 |

|
| 图 4 miR-22敲除克隆与野生型的序列比对图 用引物F1和R1对miR-22纯合缺失克隆(克隆4和克隆19)和野生型克隆分别进行PCR扩增,PCR产物的序列比对图,蓝色标记部分为敲除区域 |
RT-qPCR结果显示:miR-22纯合敲除克隆4和克隆19中,miR-22的表达量相对于野生型细胞显著降低(图 5-B),进一步证实miR-22被成功敲除。此外,miR-22缺失对小鼠胚胎干细胞的细胞形态没有显著影响(图 5-A)。并且,qPCR结果显示干性相关基因Oct4、Sox2和Nanog等表达并无明显变化(图 5-C-E)。因此,miR-22缺失并未影响小鼠胚胎干细胞的干性维持。

|
| 图 5 miR-22缺失未影响小鼠胚胎干细胞的干性维持 A:miR-22敲除并未显著影响小鼠胚胎干细胞的形态,标尺为100 μm;B:敲除克隆4和19中,miR-22的表达量显著降低,** P < 0.01 vs control;C-E:干性相关因子Oct4,Sox2和Nanog的表达并未发生显著变化 |
CRISPR/Cas9系统只需设计位点特异性的sgRNA,便可实现靶基因的敲除,具有适用面广、操作简单和节省时间等优势。该技术已经在不同物种中得到广泛应用,例如在斑马鱼[23]、果蝇[24]、小鼠[25]、拟南芥[26]、家蚕[27]、赤拟谷盗[28]等物种中实现了靶基因的精确敲除。在胚胎干细胞中,CRISPR/Cas9技术不仅可以靶向敲除目的基因,研究靶基因对维持和分化的影响,还可以在体外修复某些致病的基因突变,对转化医学和再生医学的发展有着重要的推动作用。本实验通过点突变和搭桥PCR等手段,对sgRNA载体的筛选标记进行改造,使sgRNA表达载体中的puro抗性改造为neor抗性,以满足在具有特定抗性的胚胎干细胞中进行基因敲除时药物筛选的需求。
miR-22在不同类型的细胞中具有不同的生物学功能,其调控机理的研究主要集中在肿瘤细胞、心肌细胞中。然而,先前很多生物信息学分析都暗示miR-22在小鼠胚胎干细胞的维持和分化中可能起到某些调控作用。Boyer等[29]分析RFAM数据库,发现在人胚胎干细胞中Sox2和Nanog结合在miR-22的启动子区域;Lin[30]课题组的RNA-seq数据显示,在小鼠胚胎干细胞分化的过程中,miR-22的表达量显著升高;Stadler等[31]miRNA microarray实验显示miR-22在人类胚胎干细胞分化过程中表达量有所升高;Houbaviy等[32]Northern数据也表明,miR-22在RA诱导小鼠胚胎干细胞分化过程中其表达量显著提高。此外,很多功能实验也提示miR-22参与调控谱系分化和命运决定:miR-22通过抑制TET2促进造血干细胞的自我更新和转分化[33];miR-22通过靶向HDAC6参与调控间充质干细胞的成骨分化[34];miR-22通过抑制MECP2促进胚胎干细胞向平滑肌分化[13]。上述证据都提示miR-22在小鼠胚胎干细胞中可能起到某些调控作用。然而,先前研究都是通过RNA干扰的方法进行功能分析,本实验利用CRISPR/Cas9技术构建miR-22敲除的小鼠胚胎干细胞,发现miR-22缺失并未影响胚胎干细胞的细胞形态和干性相关基因的表达,其对小鼠胚胎干细胞分化的调控机理还有待进一步分析。综上所述,本实验初步探索了miR-22在小鼠胚胎干细胞中的调控作用,对于丰富miRNA对干性维持、谱系分化和命运决定调控作用的研究有着重要的意义。
4 结论本实验通过点突变和搭桥PCR等手段,获得具有neor抗性的sgRNA表达载体,并成功构建了用于miR-22敲除的sgRNA载体。随后,利用CRISPR/Cas9技术获得miR-22敲除的小鼠胚胎干细胞,形态学观察及qPCR结果显示miR-22缺失并未影响细胞形态和干性相关基因的表达,初步探索了miR-22在小鼠胚胎干细胞中的调控作用。
| [1] |
Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells[J]. Proc Natl Acad Sci USA, 1981, 78(12): 7634-7638. DOI:10.1073/pnas.78.12.7634 |
| [2] |
Martello G, Smith A. The nature of embryonic stem cells[J]. Annu Rev Cell Dev Biol, 2014, 30: 647-75. DOI:10.1146/annurev-cellbio-100913-013116 |
| [3] |
Hackett JA, Surani MA. Regulatory principles of pluripotency:from the ground state up[J]. Cell Stem Cell, 2014, 15(4): 416-430. |
| [4] |
Huang G, Ye S, Zhou X, et al. Molecular basis of embryonic stem cell self-renewal:from signaling pathways to pluripotency network[J]. Cell Mol Life Sci, 2015, 72(9): 1741-1757. DOI:10.1007/s00018-015-1833-2 |
| [5] |
Chen T, Dent SY. Chromatin modifiers and remodellers:regulators of cellular differentiation[J]. Nat Rev Genet, 2014, 15(2): 93-106. DOI:10.1038/nrg3607 |
| [6] |
Gurtan AM, Sharp PA. The role of miRNAs in regulating gene expression networks[J]. J Mol Biol, 2013, 425(19): 3582-600. DOI:10.1016/j.jmb.2013.03.007 |
| [7] |
Ling B, Wang GX, Long G, et al. Tumor suppressor miR-22 suppresses lung cancer cell progression through post-transcriptional regulation of ErbB3[J]. J Cancer Res Clin Oncol, 2012, 138(8): 1355-1361. DOI:10.1007/s00432-012-1194-2 |
| [8] |
Tang Y, Liu X, Su B, et al. microRNA-22 acts as a metastasis suppressor by targeting metadherin in gastric cancer[J]. Mol Med Rep, 2015, 11(1): 454-460. DOI:10.3892/mmr.2014.2682 |
| [9] |
Xin M, Qiao Z, Li J, et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase:evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer[J]. Oncotarget, 2016, 7(28): 44252-44265. |
| [10] |
Guo S, Bai R, Liu W, et al. miR-22 inhibits osteosarcoma cell proliferation and migration by targeting HMGB1 and inhibiting HMGB1-mediated autophagy[J]. Tumour Biol, 2014, 35(7): 7025-7034. DOI:10.1007/s13277-014-1965-2 |
| [11] |
Li G, Wang G, Ma L, et al. miR-22 regulates starvation-induced autophagy and apoptosis in cardiomyocytes by targeting p38alpha[J]. Biochem Biophys Res Commun, 2016, 478(3): 1165-1172. DOI:10.1016/j.bbrc.2016.08.086 |
| [12] |
Ji D, Li B, Shao Q, et al. MiR-22 Suppresses BMP7 in the Development of Cirrhosis[J]. Cell Physiol Biochem, 2015, 36(3): 1026-1036. DOI:10.1159/000430276 |
| [13] |
Zhao H, Wen G, Huang Y, et al. MicroRNA-22 regulates smooth muscle cell differentiation from stem cells by targeting methyl CpG-binding protein 2[J]. Arterioscler Thromb Vasc Biol, 2015, 35(4): 918-929. DOI:10.1161/ATVBAHA.114.305212 |
| [14] |
Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea[J]. Science, 2010, 327(5962): 167-170. DOI:10.1126/science.1179555 |
| [15] |
Marraffini LA, Sontheimer EJ. CRISPR interference:RNA-directed adaptive immunity in bacteria and archaea[J]. Nat Rev Genet, 2010, 11(3): 181-190. DOI:10.1038/nrg2749 |
| [16] |
Mei Y, Wang Y, Chen H, et al. Recent Progress in CRISPR/Cas9 Technology[J]. J Genet Genomics, 2016, 43(2): 63-75. DOI:10.1016/j.jgg.2016.01.001 |
| [17] |
Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression[J]. Cell, 2013, 152(5): 1173-1183. DOI:10.1016/j.cell.2013.02.022 |
| [18] |
Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity[J]. Cell, 2013, 154(6): 1380-1389. DOI:10.1016/j.cell.2013.08.021 |
| [19] |
Wei C, Liu J, Yu Z, et al. TALEN or Cas9 - rapid, efficient and specific choices for genome modifications[J]. J Genet Genomics, 2013, 40(6): 281-289. DOI:10.1016/j.jgg.2013.03.013 |
| [20] |
Cai M, Yang Y. Targeted genome editing tools for disease modeling and gene therapy[J]. Curr Gene Ther, 2014, 14(1): 2-9. DOI:10.2174/156652321402140318165450 |
| [21] |
Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9[J]. Science, 2014, 346(6213): 1258096. DOI:10.1126/science.1258096 |
| [22] |
Wang T, Wei JJ, Sabatini DM, et al. Genetic screens in human cells using the CRISPR-Cas9 system[J]. Science, 2014, 343(6166): 80-84. DOI:10.1126/science.1246981 |
| [23] |
Xiao A, Zhang B. Generation of targeted genomic deletions through CRISPR/Cas system in Zebrafish[J]. Methods Mol Biol, 2016, 1451: 65-79. DOI:10.1007/978-1-4939-3771-4 |
| [24] |
Bassett AR, Tibbit C, Ponting CP, et al. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system[J]. Cell Rep, 2014, 6(6): 1178-1179. DOI:10.1016/j.celrep.2014.03.017 |
| [25] |
Wang H, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J]. Cell, 2013, 153(4): 910-918. DOI:10.1016/j.cell.2013.04.025 |
| [26] |
Ryder P, McHale M, Fort A, et al. Generation of stable nulliplex autopolyploid lines of Arabidopsis thaliana using CRISPR/Cas9 genome editing[J]. Plant Cell Rep, 2017, 36(6): 1005-1008. DOI:10.1007/s00299-017-2125-0 |
| [27] |
Wei W, Xin H, Roy B, et al. Heritable genome editing with CRISPR/Cas9 in the silkworm, Bombyx mori[J]. PLoS One, 2014, 9(7): e101210. DOI:10.1371/journal.pone.0101210 |
| [28] |
Gilles AF, Schinko JB, Averof M. Efficient CRISPR-mediated gene targeting and transgene replacement in the beetle Tribolium castaneum[J]. Development, 2015, 142(16): 2832-2839. DOI:10.1242/dev.125054 |
| [29] |
Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells[J]. Cell, 2005, 122(6): 947-956. DOI:10.1016/j.cell.2005.08.020 |
| [30] |
Gangaraju VK, Lin H. MicroRNAs:key regulators of stem cells[J]. Nat Rev Mol Cell Biol, 2009, 10(2): 116-125. DOI:10.1038/nrm621 |
| [31] |
Stadler B, Ivanovska I, Mehta K, et al. Characterization of microRNAs involved in embryonic stem cell states[J]. Stem Cells Dev, 2010, 19(7): 935-950. DOI:10.1089/scd.2009.0426 |
| [32] |
Houbaviy HB, Murray MF, Sharp PA. Embryonic stem cell-specific MicroRNAs[J]. Dev Cell, 2003, 5(2): 351-358. DOI:10.1016/S1534-5807(03)00227-2 |
| [33] |
Song SJ, Ito K, Ala U, et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation[J]. Cell Stem Cell, 2013, 13(1): 87-101. DOI:10.1016/j.stem.2013.06.003 |
| [34] |
Huang S, Wang S, Bian C, et al. Upregulation of miR-22 promotes osteogenic differentiation and inhibits adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells by repressing HDAC6 protein expression[J]. Stem Cells Dev, 2012, 21(13): 2531-40. DOI:10.1089/scd.2012.0014 |