




长末端重复序列(Long terminal repeat,LTR)反转录转座子是真核生物基因组中普遍存在的一类可移动的DNA序列,它们以RNA为媒介,通过“复制-粘贴”的形式在基因组中不断自我扩增。LTR反转录转座子的插入和删除对基因组的进化和物种的环境适应能力都具有重要的意义[1],此外,LTR反转录转座子、基因组重复和多倍化是导致植物基因组扩增和收缩的三个主要原因[2, 3]。LTR反转录转座子根据内部编码基因(gag,pol和int)排列顺序的不同,分为Copia和Gypsy两类主要的超家族[4]。LTR末端不编码蛋白质,但包含转录的起始信号和终止信号,内部编码区gag基因编码的蛋白负责反转录转座子RNA的成熟和包装,pol基因编码反转录酶和RNAse H,INT(Integrase)基因编码整合酶[4]。
LTR反转录转座子的插入除了可以导致基因组的膨胀外,更重要的是影响插入位点及其相邻基因的表达[5, 6]。LTR反转录转座子能够完成自我转录是因为其本身含有转录所需的调控元件。当LTR反转录转座子插入到基因编码区就可能导致该基因转录成不完整RNA序列,进而不能被翻译成完整的肽链,或者该RNA失去其调控能力;当LTR反转录转座子插入到基因附近区域时,其序列内的调控元件将会发挥作用,并影响附近基因的表达[7]。
棉纤维是纺织工业中天然纤维的主要来源。中国种植的四倍体棉花主要包括陆地棉和海岛棉两个栽培种,陆地棉丰产性好,而海岛棉不仅纤维品质优良,而且是盐碱地的主要栽培作物。随着国内纺织技术的发展和人们生活水平的提高,海岛棉的需求量也逐年增加。
四倍体海岛棉的基因组为2.57 G,大约69.11%为重复序列,其中A亚组重复序列占73.5%(1 098 Mb),D亚组重复序列占63.5%(541.6 Mb)[8, 9],而A基因组的亚洲棉与D基因组的雷蒙德氏棉中转座子的含量则分别为57.0%和68.5%[10]。在四倍体棉种转座子起源的相关研究中,发现更多的LTR反转录转座子起源于A基因组,或者说,四倍体A亚组中有更多的转座子拷贝[11, 12]。此外,雷蒙德氏棉的转座子数据库也已经公布[13]。这些数据为深入研究海岛棉LTR反转录转座子提供了绝佳的机遇。本研究首先综合多种不同的方法挖掘了海岛棉基因组中的LTR反转录转座子,然后对这些转座子进行了家族分类、周边基因的功能富集、数量分布和统计分析。本研究对海岛棉基因功能分析和基因组进化有重要的参考价值。
1 材料与方法 1.1 材料海岛棉的基因组序列、基因注释和GO注释文件均从COTTONGEN(https://www.cottongen.org/)下载。
1.2 方法 1.2.1 海岛棉LTR反转录转座子的挖掘与分类分别通过依据LTR反转录转座子结构特征的工具LTR_STRUC[14]和LTRharvest[15];依据LTR反转录转座子重复特征的工具PILER[16];以及综合性工具RepeatModeler[17]搜寻海岛棉基因组中的LTR反转录转座子。使用REPCLASS[18]软件将上述结果序列归类到相应的超家族,并将相同的超家族合并,之后再与已知重复序列数据库Repbase[19]进行进一步的合并。利用CD-HIT[20]去除合并结果中的冗余序列,得到海岛棉特异的LTR反转录转座子序列库,最后利用RepeatMasker[21]注释海岛棉基因组中的LTR反转录转座子,由同一参考序列注释到的一组序列被认为是一个家族。分析过程中,要求LTR反转录转座子序列最短为80 bp,每个LTR反转录转座子家族在基因组中有100次以上的重复拷贝。
1.2.2 海岛棉LTR反转录转座子的数量与分布通过Perl脚本从RepeatMasker结果文件中收集LTR反转录转座子在海岛棉基因组的数量和位置,并利用gff注释文件的数据收集基因的数量和位置。统计分析染色体中每100 kb范围内的LTR反转录转座子与基因的数量,并通过Circos[22]绘制分布图。
1.2.3 海岛棉LTR反转录转座子周边基因的GO富集分析查找海岛棉LTR反转录转座子上、下游20 kb范围内的基因,利用基因组GO注释文件确定这些基因的GO注释条目,并使用WEGO[23](http://wego.genomics.org.cn/)进行富集分析。
2 结果 2.1 海岛棉LTR反转录转座子的数量分析海岛棉为四倍体棉种,A亚组和D亚组各包含13条染色体。数据结果(图 1)表明海岛棉基因组中共包含2 018个100拷贝以上的LTR反转录转座子家族,其中1 930个家族共同存在于A亚组和D亚组中,84个家族为A亚组特异,4个家族为D亚组特异。A亚组共包含274 360个LTR反转录转座子拷贝,D亚组则包含209 415个拷贝,因此,LTR反转录转座子在A亚组中的拷贝要比D亚组多,这一特征对于高拷贝数的LTR反转录转座子家族表现的更为明显(图 2)。此外,从图 2还可以大致看出,一个LTR反转录转座子家族在较大的染色体上有较多的拷贝数,在较小的染色体上有较少的拷贝数,LTR反转录转座子家族特异分布在少数染色体上的情况并不明显。

|
| 图 1 海岛棉A亚组和D亚组共有和特异的LTR反转录转座子家族 |

|
| 图 2 不同拷贝数的LTR反转录转座子家族在海岛棉各染色体上的分布 不同颜色表示不同拷贝数;同一行表示相同的LTR反转录转座子家族;从上到下家族拷贝数逐渐增多 |
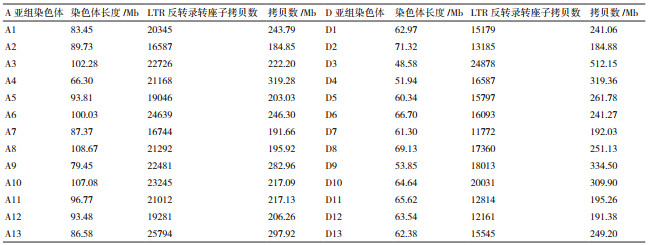
海岛棉基因组每Mb序列平均包含242.21拷贝的LTR反转录转座子,通过计算发现海岛棉基因组中每Mb染色体包含的LTR反转录转座子的拷贝数与染色体大小的皮尔森相关系数为-0.52,表现出中度负相关的关系(表 1)。
海岛棉基因组中的LTR反转录转座子整体在A亚组染色体的后端部分分布较少(4号染色体除外),而在D亚组染色体则相对分布均匀(图 3)。Copia超家族在染色体的起始端有一个明显的波峰,这一特点A亚组和D亚组类似,同时,Copia超家族在A亚组染色体的后端部分分布较少,而D亚组染色体则没有该特征。Gypsy超家族整体在染色体中部分布多,两端分布少,进一步比较A亚组和D亚组可以发现,Gypsy超家族在A亚组后半部分分布的更少。因此,A亚组染色体后端LTR反转录转座子分布较少是这两类主要超家族共同的分布特征。

|
| 图 3 LTR反转录转座子在海岛棉基因组上的分布 A:全部LTR反转录转座子;B:Copia超家族;C:Gypsy超家族;D:基因;E:染色体 |
比较海岛棉基因组中基因与LTR反转录转座子的分布发现Gypsy超家族与基因的分布呈近似反比关系,而Copia超家族与基因则没有明显的数量分布关系。
2.3 海岛棉LTR反转录转座子周边基因的富集分析分别对海岛棉A亚组与D亚组LTR反转录转座子上下游20 kb范围内的基因进行了GO富集分析。结果共涉及GO分类体系中的细胞组件、分子功能和生物过程3个大类别中的9、12和19个小类别。A亚组和D亚组LTR反转录转座子周边分别有47 979和55 880个基因具有GO注释。从图 4中可以看出两类超家族的富集情况基本相同,在细胞组件中,涉及较多的条目依次是细胞(Cell GO:0005623)、细胞组分(Cell part GO:0044464)、细胞器(Organelle GO:0043226)和大分子复合物(Macromolecular complex GO:0032991)等;在分子功能方面,主要富集在结合活性(Binding GO:0005488)和催化活性(Catalytic activity GO:0003824)等类别中;而在生物学过程中,涉及较多的条目依次是代谢过程(Metabolic process GO:0008152)、细胞过程(Cellular process GO:0009987)、生物调节(Biological regulation GO:0065007)、定位(Localization GO:0051179)、建立定位(Establishment of localization GO:0051234)和色素(Pigmentation GO:0043473)等。将二倍体亚洲棉(A组)、雷蒙德氏棉(D组)与四倍体海岛棉(AD组)进行比较,发现LTR反转录转座子周边基因的GO富集情况类似,只是在基因数量和百分比方面有差别(数据未发表)。

|
| 图 4 海岛棉LTR反转录转座子周边基因的GO富集分析 |
LTR反转录转座子是植物基因组的重要成分,是推动基因组大小变异和进化的重要因素[2]。精确而完整的LTR反转录转座子注释对研究基因组大小变异和进化具有非常重要的意义。从基因组中挖掘转座子序列的算法主要有三类:依据转座子的结构特征、依据转座子在基因组中的重复特征和依据已知转座子序列进行同源搜索,每种方法都有各自的优势和缺陷[24]。本文首先综合使用前两类算法的软件挖掘了海岛棉基因组中的转座子序列,再进一步将这些序列合并、去冗余,构建出一个海岛棉特异转座子序列数据库。最后依据该库通过RepeatMasker软件用同源搜索的方法注释海岛棉基因组中的转座子序列,从而获得了海岛棉基因组中非常完整的转座子信息。进一步的数据分析发现海岛棉基因组中每Mb染色体包含的LTR反转录转座子的拷贝数与染色体的大小具有一定的负相关性,但这一特征在其他物种中是否存在还需要进一步的研究。
LTR反转录转座子在海岛棉A亚组和D亚组的分布曲线有较大的差别(图 3),而在相同亚组内部的各染色体上则具有类似的分布曲线(A亚组4号染色体除外)。高拷贝数的LTR反转录转座子家族在海岛棉A亚组和D亚组的分布特征也具有一定的差异。此外,在转座子活性研究中发现一个Copia类转座子仅插入到海岛棉A亚组的HD1基因中,而在D亚组的HD1基因中则没有插入[25]。这些数据都表明转座子在四倍体海岛棉A亚组和D亚组中并不是完全相同的。然而,LTR反转录转座子周边基因的富集分析则表明A、D亚组之间非常类似。研究通过比较四倍体海岛棉A、D亚组LTR反转录转座子的数量与分布特征使我们对其有了更好的认识,这将为海岛棉基因组研究提供数据支持。
4 结论本研究结果表明,绝大多数LTR反转录转座子家族被海岛棉A亚组和D亚组共同拥有,同时,两个亚组也分别存在少数特异家族。海岛棉染色体的大小与LTR反转录转座子的数量有关。此外,本研究也发现在海岛棉基因组中,Gypsy超家族分布较多的位置基因分布较少,但Copia超家族的分布则与基因没有明显的关系。
| [1] |
Oliver KR, McComb JA, Greene WK. Transposable elements:powerful contributors to angiosperm evolution and diversity[J]. Genome Biol Evol, 2013, 5(10): 1886-1901. DOI:10.1093/gbe/evt141 |
| [2] |
Bennetzen JL. Transposable element contributions to plant gene and genome evolution[J]. Plant Mol Biol, 2000, 42(1): 251-269. DOI:10.1023/A:1006344508454 |
| [3] |
Vitte C, Panaud O. LTR retrotransposons and flowering plant genome size:emergence of the increase/decrease model[J]. Cytogenet Genome Res, 2005, 110(1-4): 91-107. DOI:10.1159/000084941 |
| [4] |
Wicker T, Sabot F, Hua-Van A, et al. A unified classification system for eukaryotic transposable elements[J]. Nat Rev Genet, 2007, 8(12): 973-982. DOI:10.1038/nrg2165 |
| [5] |
Kobayashi S, Goto-Yamamoto N, Hirochika H. Retrotransposon-induced mutations in grape skin color[J]. Science, 2004, 304(5673): 982. DOI:10.1126/science.1095011 |
| [6] |
Mirouze M, Reinders J, Bucher E, et al. Selective epigenetic control of retrotransposition in Arabidopsis[J]. Nature, 2009, 461(7262): 427-430. DOI:10.1038/nature08328 |
| [7] |
Domingues DS, Cruz GM, Metcalfe CJ, et al. Analysis of plant LTR-retrotransposons at the fine-scale family level reveals individual molecular patterns[J]. BMC Genomics, 2012, 13(1): 137. DOI:10.1186/1471-2164-13-137 |
| [8] |
Liu X, Zhao B, Zheng HJ, et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites[J]. Sci Rep, 2015, 5: 14139. DOI:10.1038/srep14139 |
| [9] |
Yuan D, Tang Z, Wang M, et al. The genome sequence of Sea-Island cotton(Gossypium barbadense)provides insights into the allopolyploidization and development of superior spinnable fibres[J]. Sci Rep, 2015, 5: 17662. |
| [10] |
Wang K, Huang G, Zhu Y. Transposable elements play an important role during cotton genome evolution and fiber cell development[J]. Sci China Life Sci, 2016, 59(2): 112-121. DOI:10.1007/s11427-015-4928-y |
| [11] |
Hu G, Hawkins JS, Grover CE, et al. The history and disposition of transposable elements in polyploid Gossypium[J]. Genome, 2010, 53(8): 599-607. DOI:10.1139/G10-038 |
| [12] |
Hawkins JS, Kim H, Nason JD, et al. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium[J]. Genome Res, 2006, 16(10): 1252-1261. DOI:10.1101/gr.5282906 |
| [13] |
Xu Z, Liu J, Ni W, et al. GrTEdb:the first web-based database of transposable elements in cotton(Gossypium raimondii)[J]. Database(Oxford), 2017, 2017(1). |
| [14] |
McCarthy EM, McDonald JF. LTR_STRUC:a novel search and identification program for LTR retrotransposons[J]. Bioinformatics, 2003, 19(3): 362-367. DOI:10.1093/bioinformatics/btf878 |
| [15] |
Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons[J]. BMC Bioinformatics, 2008, 9(1): 1-14. |
| [16] |
Edgar RC, Myers EW. PILER:identification and classification of genomic repeats[J]. Bioinformatics, 2005, 21(Suppl 1): i152-i158. DOI:10.1093/bioinformatics/bti1003 |
| [17] |
Huda A, Jordan IK. Analysis of transposable element sequences using CENSOR and RepeatMasker[J]. Methods Mol Biol, 2009, 537(537): 323-336. |
| [18] |
Feschotte C, Keswani U, Ranganathan N, et al. Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes[J]. Genome Biol Evol, 2009, 1(1): 205-220. |
| [19] |
Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes[J]. Mob DNA, 2015, 6(1): 11. DOI:10.1186/s13100-015-0041-9 |
| [20] |
Fu L, Niu B, Zhu Z, et al. CD-HIT:accelerated for clustering the next-generation sequencing data[J]. Bioinformatics, 2012, 28(23): 3150-3152. DOI:10.1093/bioinformatics/bts565 |
| [21] |
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences[J]. Curr Protoc Bioinformatics, 2009, Chapter 4: 4-10. |
| [22] |
Naquin D, D'Aubenton-Carafa Y, Thermes C, et al. CIRCUS:a package for Circos display of structural genome variations from paired-end and mate-pair sequencing data[J]. BMC Bioinformatics, 2014, 15(1): 198. DOI:10.1186/1471-2105-15-198 |
| [23] |
Ye J, Fang L, Zheng H, et al. WEGO:a web tool for plotting GO annotations[J]. Nucleic Acids Res, 2006, 34(Web Server issue): W293-W297. |
| [24] |
Lerat E. Identifying repeats and transposable elements in sequenced genomes:how to find your way through the dense forest of programs[J]. Heredity(Edinb), 2010, 104(6): 520-533. |
| [25] |
Cao Y, Jiang Y, Ding M, et al. Molecular characterization of a transcriptionally active Ty1/copia-like retrotransposon in Gossypium[J]. Plant Cell Rep, 2015, 34(6): 1037-1047. DOI:10.1007/s00299-015-1763-3 |