




2. 天津市农业质量标准与检测技术研究所,天津 300381;
3. 天津农学院,天津 300382;
4. 中国农业科学院生物技术研究所,北京 100081
2. Tianjin Institute of Agricultural Quality Standard and Testing Technology, Tianjin Academy of Agricultural Sciences, Tianjin 300381;
3. Tianjin Agricultural University, Tianjin 300384;
4. Biotechnology Research Institute, Chinese Academy of Agricultural Sciences, Beijing 100081
截止到2015年,全球共有40个国家批准转基因作物用于粮食或饲料,涉及26种转基因作物的363个转基因转化事件[1],累计种植面积达20亿hm2。伴随转基因技术的发展和人们生活水平的提高,转基因作物及其衍生食品的安全问题受到社会强烈关注和争议[2-3]。世界各个国家、组织和地区,都对转基因作物的上市和推广规定了明确的政策和法规[2, 4],对其开展严格的生物安全评价。
表达载体、目的基因整合情况、外源插入序列表达情况等分子特征是转基因生物安全评价的重要内容,其中转基因作物中插入外源基因的拷贝数鉴定是关键参数[5]。以往的拷贝数检测方法有Southern杂交[6]、实时荧光PCR[7-9]、普通PCR方法[10]等,但在实验步骤、标准物质要求等方面存在一定的局限性。微流滴数字PCR是新一代PCR技术,其原理是将PCR体系通过微流滴的形式分配成20 000甚至更多个微滴,经PCR扩增后进行信号阅读获得阳性微滴和阴性微滴的数量,再经过泊松分布的计算原理获得样品中靶标基因浓度。本研究应用微流滴数字PCR方法鉴定转基因水稻目的基因拷贝数,并与Southern杂交结果进行比较,以探讨微流滴数字PCR技术在转基因作物目的基因拷贝数的应用性。
1 材料与方法 1.1 材料转Cry1Ab基因抗虫转基因水稻及其非转基因受体由本实验室保存。转基因水稻具有二化螟、三化螟、稻纵卷叶螟以及大螟的抗性,外源片段序列共6 600 bp,包括Ubi启动子、Cry1Ab目的基因、T-nos终止子、T-35S部分序列等。
1.2 方法 1.2.1 DNA提取ddPCR试验用DNA模板使用新型植物基因组DNA提取试剂盒(天根,DP-320)提取,Southern杂交用DNA模板使用CTAB大量植物基因组DNA快速提取试剂盒(北京艾德莱,DN1404)。
1.2.2 引物筛选及设计试验用内标准基因、目的基因、转化体特异性序列引物和探针序列,见表 1。

引物SPS和TJR使用普通PCR反应体系(仪器为ABI,Veriti96 PCR仪)。总体积为25 µL,包括2×Gotaq Green Master Mix(Promega公司)12.5 µL,25 mg/L DNA模板2.0 µL,10 µmol/L上下游引物各1.0 µL(SPS引物为SPS-F和SPS-R各1.0 µL,TJR引物为复合体系,其中TJR-F1 2.0 µL、TJR-R1 1.0 µL、TJR-R2 1.0 µL),用ddH2O补足25 µL。PCR反应程序:94℃预变性5 min;94℃变性30 s,58℃退火30 s,72℃延伸40 s,35次循环;72℃延伸5 min。反应结束后取出PCR管,对PCR扩增产物进行电泳检测(2%琼脂糖)。
1.2.4 数字PCR方法引物PLD和Bt使用微流滴数字PCR反应体系(仪器为Bio-Rad,QX100微流滴数字PCR仪)。总体积为20 µL,包括2×ddPCR Supermix for probes(Bio-Rad)10 µL,25 mg/L DNA模板(适当稀释后)2.0 µL,10 µmol/L上下游引物和探针各1.0 µL,用ddH2O补足20 µL。将20 μL反应体系小心转移至微滴发生卡,同时于微滴发生卡相应位置加入微滴发生专用油70 μL,盖密封垫,放入droplet generator中生成微滴;小心吸取40 μL生成的微滴,转移至半裙边96孔PCR反应板中,加盖铝膜,置于封膜仪中170℃热封。将热封后的反应板放入PCR仪(Analytik Jena,EasyCycler 96 PCR仪)扩增,反应程序:95℃预变性10 min;94℃变性30 s,60℃退火1 min,共进行40次循环;98℃,10 min;PCR仪升降速度设置为2℃/s。扩增结束后,将反应板放入droplet reader,根据软件(QuantaSoft Software)提示设定参数及数据分析。
1.2.5 Southern杂交方法探针制备PCR扩增使用CryIA(b)引物(仪器为ABI,Veriti96 PCR仪)。总体积为50 µL,包括2×Gotaq Master Mix(Promega公司)25 µL,25 mg/L DNA模板4.0 µL,10 µmol/L上下游引物各2.0 µL,用ddH2O补足50 µL。PCR反应程序为:94℃预变性5 min;94℃变性30 s,58℃退火30 s,72℃延伸60 s,共进行35次循环;72℃延伸5 min。反应结束后取出PCR管,对PCR扩增产物进行电泳检测(1%琼脂糖)及地高辛探针制备(Roche公司)。基因组DNA模板酶切体系总体积为800 µL,包括10×Buffer 80 µL,内切酶40 µL,基因组DNA 10 µg。探针和酶切产物经电泳、转膜、杂交等后续步骤获得Southern杂交结果。
2 结果 2.1 数字PCR方法结果 2.1.1 DNA提取结果分析分别准备转基因水稻及其受体材料各4份,取单粒进行萌发,采用试剂盒法提取基因组DNA,使用NanoDrop ND-1000微量分光光度计测量DNA浓度、A260/A280、A260/A230等指标。结果表明(表 2),提取的DNA浓度在96.8-123.5之间,A260/A280均在1.8-2.0之间,说明纯度较高。

对提取的基因组DNA进行水稻内标准SPS基因的PCR扩增,电泳结果(图 1)显示,8个水稻材料均扩增出277 bp大小的预期片段,条带清晰单一,说明提取的基因组DNA满足PCR的要求。

|
| 图 1 SPS基因PCR扩增结果 M:DL2000 Marker;1:空白对照;2-5:转基因水稻;4-9:受体材料 |
在进行数字PCR鉴定拷贝数之前,需对材料进行纯合性鉴定。依据转化体特异性序列信息,设计纯合性鉴定复合PCR引物TJR(表 1)。其中TJR-F1/R1靶标为水稻基因组序列,TJR-F1/R2靶标为转基因水稻转化体特异性序列;纯合阴性材料仅能扩增出376 bp大小的TJR-F1/R1扩增产物,纯合阳性材料仅能扩增出741 bp大小的TJR-F1/R2扩增产物,杂合材料能扩增出TJR-F1/R1和TJR-F1/R2两种扩增产物。复合PCR结果显示,本试验选择的转基因水稻阴性和阳性材料均为纯合材料(图 2)。

|
| 图 2 转基因水稻纯合度鉴定结果 M:DL2000 Marker ;1:空白对照;2:混样对照;3-6:阴性材料;7-10:阳性材料 |
以4份转基因水稻基因组DNA为模板,应用微流滴数字PCR技术扩增内标准基因PLD和目的基因Cry1Ab。结果显示,生成微滴总数量在13 000-16 000之间(图 3);阳性微滴和阳性微滴荧光信号分离界限明显(图 4);4个重复的测得目的/内标准基因的比值分别为674/662、516/541、737/759、528/571(图 5),均值为0.97≈1。本试验采用的内标准基因的拷贝数为1,试验材料经验证为纯合材料,因此确定目的外源基因Cry1Ab在该转基因水稻品系中的拷贝数为1。

|
| 图 3 微流滴数字PCR微滴数量结果 |

|
| 图 4 微流滴数字PCR微滴荧光信号 |

|
| 图 5 微流滴数字PCR测定PLD和Cry1Ab拷贝数浓度结果图 |
根据外源插入序列全长及探针序列信息,筛选出无酶切位点的限制性内切酶共39种(表 3);选择其中Nco Ⅰ、Sph Ⅰ、Sac Ⅰ、BamH Ⅰ、Hind Ⅲ、Pst Ⅰ和Mse Ⅰ共7种内切酶用于Southern杂交试验(表 3中底纹标识)。
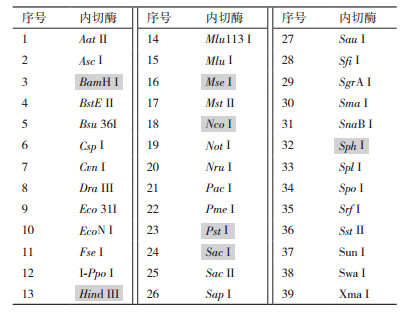
应用上述7种内切酶,对转基因水稻基因组DNA酶切,结果见图 6。从图中可见,酶切后泳道呈弥散状,无明显条带,说明基因组DNA酶切效果较好。用引物Cry1Ab扩增产物为杂交探针,对酶切产物进行Southern-blotting试验。杂交结果(图 7)显示,转基因水稻基因组DNA经7种内切酶酶切后,探针杂交信号均为1个,说明目的基因的拷贝数为1。

|
| 图 6 转基因抗虫水稻酶切结果 1:Nco Ⅰ;2:Sph Ⅰ;3:Sac Ⅰ;4:BamH Ⅰ;5:Hind Ⅲ;6:Pst Ⅰ;7:Mse Ⅰ;8:Marker |

|
| 图 7 转基因抗虫水稻Southern杂交结果图 M:Marker;1:阳性质粒;2:Nco Ⅰ;3:Sph Ⅰ;4:Sac Ⅰ;5:BamH Ⅰ;6:Hind Ⅲ;7:Pst Ⅰ;8:Mse Ⅰ |
Southern杂交是最经典的外源基因拷贝数鉴定的技术[11],依据靶标探针与酶切基因组DNA杂交信号的数量对外源基因的拷贝数进行鉴定,试验步骤包括探针制备、基因组DNA酶切、转膜、杂交等。燕树锋等[12]应用Southern杂交方法鉴定了转CrylAc-w基因抗虫玉米目的基因的插入位点。Southern杂交技术具有灵敏度高、无需已知拷贝数的内标准基因等优点,现在仍然被众多实验室应用。但同时,由于整个操作流程较费事费力且需要大量的DNA模板,因此近年来该技术应用范围越来越小,逐步被其他技术取代。
宋锋等[10]以转基因烟草植株为对象,针对已知单拷贝的烟草内标准基因RNR2和待测拷贝数外源基因NPTⅡ设计了相同退火温度的PCR引物,通过软件计算复合定性PCR的电泳产物的灰度比,确定了7株不同转基因烟草中外源基因NPTⅡ的拷贝数。该方法具有过程简单、高效、成本低的技术优点,但同时对引物退火温度、扩增效率、PCR产物大小等提出严格条件,以避免在成像过程中造成误差。
实时荧光PCR技术被应用于小麦、棉花、水稻、大豆和玉米等转基因作物外源基因拷贝数鉴定。杨晓杰等[13]基于SYBR Green定量PCR技术的PfaffI法,比较了农杆菌介导法、基因枪轰击法和花粉管通道法等3种常用转化方法获得的转基因棉花中外源基因拷贝数的差异,结果表明实时荧光PCR方法满足高通量地检测大规模转化体转基因拷贝数;李淑洁等[9]采用SYBR Green real time PCR方法检测7株转基因小麦中外源半夏凝集素基因的拷贝鉴定出单拷贝、2拷贝、3拷贝和4拷贝植株;王育花等[14]利用SYBR Green Ⅰ荧光定量实时PCR法检测了转大麦烟酰胺合成酶基因水稻中外源基因拷贝数,鉴定出1、2、3、4和7个拷贝;杨冬燕等[15]利用TaqMan定量PCR技术鉴定了转基因大豆RRS和转基因玉米Bt176中外源基因拷贝数,结果为Bt176玉米中CrylA(b)基因的整合拷贝数分别为2.69,3.43和2.83,平均为2.9,RRS大豆中EPSPS基因拷贝数分别为1.08、1.01和0.96,平均为1.0。上述方法表明实时荧光PCR方法具有较强的适用性和外源插入拷贝数分辨率。但是该技术必须使用标准物质(或标准质粒)绘制标准曲线,对靶标基因和已知拷贝数的内标准基因进行浓度测定。
微流滴数字PCR是继定性PCR、实时荧光PCR之后的第三代PCR技术。微流滴数字PCR技术具有绝对定量、无需构建标准曲线等技术特点,已应用于转基因产品成分精准定量等研究中[16-18]。本研究应用这一技术对转基因水稻内标准基因PLD(在水稻基因组中为单拷贝[19-20])和目的基因Cry1Ab浓度进行绝对定量检测,通过计算Cry1Ab/PLD,获得转基因水稻目的基因Cry1Ab的插入拷贝数[21-22]。与普通PCR和实时荧光PCR方法相比,微流滴数字PCR无需通过构建标准曲线来定量靶标基因和内标准基因的拷贝数浓度,因此适用性更强。
4 结论本研究应用微流滴数字PCR技术鉴定转基因抗虫水稻中外源目的基因Cry1Ab在基因组的插入拷贝数。对4个转基因纯合单株进行检测,结果分别1.02、0.95、0.97、0.92,因此鉴定为单拷贝插入。该结果与经典方法Southern杂交鉴定结果一致。
| [1] |
James C. Brief 51:20th Anniversary(1996 to 2015)of the Global Commercialization of Biotech Crops and Biotech Crop Highlights in 2015[M]. Ithaca, NY, USA: ISAAA, 2015.
|
| [2] |
Goodman RE. Biosafety:evaluation and regulation of genetically modified(GM)crops in the United States[J]. Journal of Huazhong Agricultural University, 2014, 33(6): 109-114. |
| [3] |
Sanvido O, Romeis J, Gathmann A, et al. Evaluating environmental risks of genetically modified crops:ecological harm criteria for regulatory decision-making[J]. Environmental Science & Policy, 2012, 15(1): 82-91. |
| [4] |
Zepeda J, Cohen J. Biosafety regulation of genetically modified orphan crops in developing countries: A way forward[M]//regulating agricultural biotechnology: Economics and policy, US Springer, 2006: 509-533.
|
| [5] |
农业部办公厅. 农业转基因生物安全评价指南[S]: 北京: 2017.
|
| [6] |
华志华, 等. 基因枪转化获得的转基因水稻中外源基因整合与表达规律研究[J]. 遗传学报, 2001, 28(11): 1012-1018. |
| [7] |
杨立桃, 等. 利用实时荧光定量PCR方法分析转基因水稻外源基因拷贝数[J]. 中国食品卫生杂志, 2005(2): 140-144. |
| [8] |
孔庆然, 等. 转基因猪中外源基因拷贝数和整合位点的研究[J]. 生物化学与生物物理进展, 2009(12): 1617-1625. |
| [9] |
李淑洁, 张正英. REAL-TIME PCR方法测定转基因小麦中外源基因拷贝数[J]. 中国生物工程杂志, 2010, 30(3): 90-94. |
| [10] |
宋锋, 孙敏, 罗克明. 一种基于PCR技术鉴定单拷贝转基因烟草的方法[J]. 中国生物工程杂志, 2010, 30(4): 83-88. |
| [11] |
罗滨, 陈永康, 王莹. 植物外源基因拷贝数及插入位点的检测方法与技术[J]. 河南师范大学学报:自然科学版, 2012, 40(6): 111-116. |
| [12] |
燕树锋, 铁双贵, 岳润清, 等. 转Cry1Ac-w基因抗虫玉米的获得及分子鉴定[J]. 华北农学报, 2015(5): 41-44. DOI:10.7668/hbnxb.2015.05.007 |
| [13] |
杨晓杰, 刘传亮, 张朝军, 等. 不同转化方法获得的转基因棉花外源基因拷贝数分析[J]. 农业生物技术学报, 2011, 19(2): 221-229. |
| [14] |
王育花, 等. 利用实时荧光定量PCR法检测转基因水稻外源基因拷贝数的研究[J]. 生命科学研究, 2007(4): 301-305. |
| [15] |
杨冬燕, 郭良让, 杨小柯, 等. 利用TaqMan定量PCR技术不依赖参比内源基因测定转基因植物外源基因拷贝数[J]. 中国卫生检验杂志, 2008, 18(6): 992-996. |
| [16] |
Dobnik D, Spilsberg B, Bogožalec Košir A, et al. Multiplex quantification of 12 European Union authorized genetically modified maize lines with droplet digital polymerase chain reaction[J]. Analytical Chemistry, 2015, 87(16): 8218-8226. DOI:10.1021/acs.analchem.5b01208 |
| [17] |
Gerdes L, Iwobi A, Busch U, et al. Optimization of digital droplet polymerase chain reaction for quantification of genetically modified organisms[J]. Biomol Detect Quantif, 2016, 7: 9-20. DOI:10.1016/j.bdq.2015.12.003 |
| [18] |
Zhu P, Wang C, Huang K, et al. A novel pretreatment-free duplex chamber digital PCR detection system for the absolute quantitation of GMO samples[J]. Int J Mol Sci, 2016, 17(3): 402. DOI:10.3390/ijms17030402 |
| [19] |
Wang C, Jiang L, Rao J, et al. Evaluation of four genes in rice for their suitability as endogenous reference standards in quantitative PCR[J]. J Agric Food Chem, 2010, 58(22): 11543-11547. DOI:10.1021/jf102092c |
| [20] |
Wu G, Wu Y, Nie S, et al. Real-time PCR method for detection of the transgenic rice event TT51-1[J]. Food Chemistry, 2010, 119(1): 417-422. DOI:10.1016/j.foodchem.2009.08.031 |
| [21] |
Dalmira FU, Melina PU, Joseé-Benigno VT, et al. Development, optimization, and evaluation of a duplex droplet digital PCR assay to quantify the T-nos/hmg copy number ratio in genetically modified maize[J]. Analytical Chemistry, 2015, 88(1): 812-819. |
| [22] |
Jiang Y, Hu JY, Yang LT. Estimating the exogenous genes copy number of genetically modified organisms by droplet digital PCR[J]. J Agric Biotechnol, 2014, 22(10): 1298-1305. |