



木通红喀木虱的寄主植物是木通、白木通等木通科植物, 它广泛分布于我国及、日韩等邻国[1-5]。木通红喀木虱不仅危害植物, 其携带传播的植物病毒等更是危害严重[6-10]。为保护木通等药用植物资源, 了解木通红喀木虱生物学性质, 寻找其生理生化特性, 对其线粒体基因组的研究尤为重要。然而, 我国对木虱的研究起步较晚, 且进展缓慢, 远落后于许多国家。
本研究基于PCR扩增、基因克隆等技术通过拼接得到木通红喀木虱线粒体全长, 并获得其结构及碱基含量, 蛋白质基因及密码子, 核糖体RNA和转运RNA基因等情况。利用DNAStar 6.0、Mega 6.0软件获得序列的总长、碱基含量、密码子使用、氨基酸组成等统计分析。通过木虱线粒体基因组序列的研究, 能够了解其在木虱科及同翅目系统发育中的地位, 及其与同翅目其他昆虫的系统发育关系[11-13]。线粒体基因的诸多优势, 如保守性高、变异较快、有较快的进化速率、母系遗传及遗传过程中不发生重组等[14-18], 使得在研究木虱系统发育与进化关系、亲缘关系、系统地理学研究和物种鉴定等问题上有重大意义。此外, 由于该线粒体基因组序列的测定为物种的鉴定提供重要分子支持, 所以在害虫预警, 预防生物入侵上均有重大意义。
1 材料与方法 1.1 材料本研究所用的木通红喀木虱样品2017年7-8月采集于江西省九江, 现保存在南昌大学螨类实验室, -20℃冰箱保藏。
1.2 方法 1.2.1 DNA提取、PCR扩增与产物纯化挑取3-4龄木通红喀木虱幼虫进行总DNA提取。方法参照Qiagen DNeasy Tissue Kit说明书提取总DNA。
根据引物设计原则, 参考mtDNA的通用引物[19-20]和白粉虱Trialeurodes vaporariorum的长片段引物以及其他相关种类的全线粒体基因组测序的PCR引物[21-22], 尽量在保守区域进行引物的设计, 同时考虑引入简并位点增加引物通用性和引物步移的方法来设计引物, 同时还要考虑各设计PCR引物要覆盖线粒体基因组全序列。
首先采用昆虫线粒体基因(cox1[19]、cox3[23]、cob[19]、rrnS[19]、rrnL[19]和nad5[23])的5对通用引物进行PCR扩增, 得到了木通红喀木虱线粒体cox1、cox3、nad5、cob、12S rRNA和16S rRNA 6个基因的部分序列。然后再根据所上述基因测序结果, 设计出相应的特异性引物(表 1)。然后根据上述扩增长片段引物用LaTaq DNA聚合酶酶(TaKaLa)扩增, 得到4个长片段。
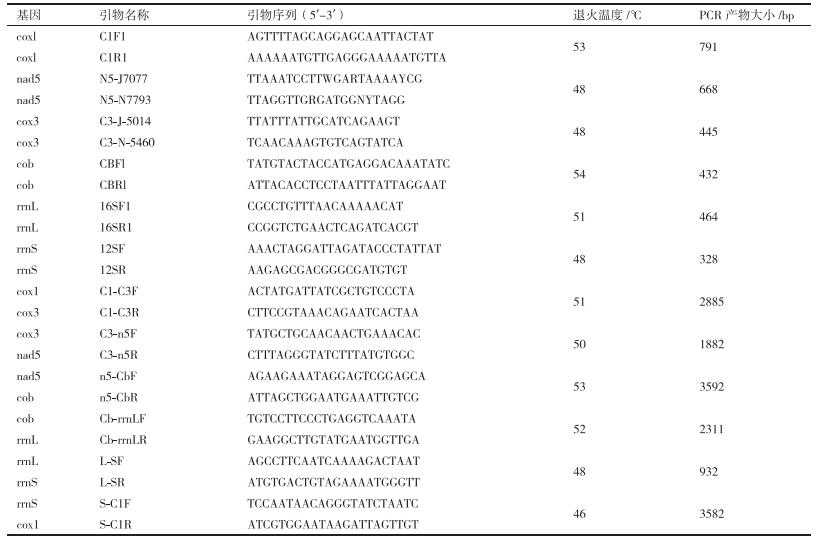
Long-PCR反应体系及扩增程序见表 1。对于小于1 kb的扩增片段, 采用2×Mix Taq DNA聚合酶; 而对于扩增1 kb以上的大片段, 则利用LA Taq DNA聚合酶。PCR产物纯化参照上海生工SanProp柱式DNA胶回收试剂盒说明进行。
1.2.2 克隆、测序与测序结果拼接PCR扩增出单一条带的PCR产物直接测序或进行克隆。对于扩增出非特异条带的PCR产物经割胶回收试剂盒(上海生工)回收后, 首先在Nanovue超微量紫外分光光度计上测定浓度及纯度。然后将PCR产物纯化, 纯化后的目的基因连接至pGEM-T Easy载体上, 将重组子转化到E.coli DH5α。最后通过蓝白斑筛选法挑取1-2个阳性克隆菌, 送至上海生工进行双向测序。
利用DANStar中的SeqMan软件对已测的木通红喀木虱mtDNA中的短片段和长片段进行拼接和校正。在原始测序峰图的基础上对拼接结果进行人工校正, 去除空间隔区和校正错误碱基。
1.2.3 线粒体基因组基因注释与基因组成情况的分析利用NCBI网站上的ORF软件(http://www.ncbi.nlm.nih.gov/projects/gorf/)注释蛋白质编码基因, 并通过木虱科的昆虫全线粒体基因组序列比对方式及MITOS WebServer程序鉴定木通红喀木虱线粒体基因组tRNA和rRNA。
线粒体基因组注释完后利用DNAStar 6.0软件包中的Editseq及Mega 6.0进行序列的总长、碱基含量、密码子使用、氨基酸组成等统计分析。
2 结果 2.1 木通红喀木虱线粒体基因组分析 2.1.1 基因组的结构分析木通红喀木虱全线粒体基因组长度为14 832 bp(图 1)(NCBI登录号为NC_027087.1)。此线粒体基因组总共编码37个基因, 包括13个蛋白质编码基因、22个转运RNA基因, 2个核糖体RNA基因, 其中L链编码14个基因, 包括4个蛋白质基因:nad1、nad4、nad4L、nad5;8个转运RNA基因:trnF、trnH、trnP、trnL1、trnV、trnQ、trnC、trnY; 2个核糖体RNA基因:rrnL(16S核糖体RNA基因)和rrnS(12S核糖体RNA基因)。剩余的23个基因则由H链编码, 这些蛋白质编码基因、转运RNA基因和核糖体RNA基因的位置以及结构都很保守。

|
| 图 1 木通红喀木虱线粒体基因组结构图示 |
如表 2所示, 木通红喀木虱全线粒体基因组的A+T含量为72.04%。蛋白质编码基因的A+T含量略有降低为70.50%, tRNA基因A+T含量为74.69%, rRNA基因的A+T含量最高为77.06%。同时, 还可得到G、C碱基含量偏斜度远高于A、T碱基的结果, AT-skew值为0.059且GC-skew值为-0.284, 其中T碱基含量高于A碱基含量, C碱基含量高于G碱基含量。
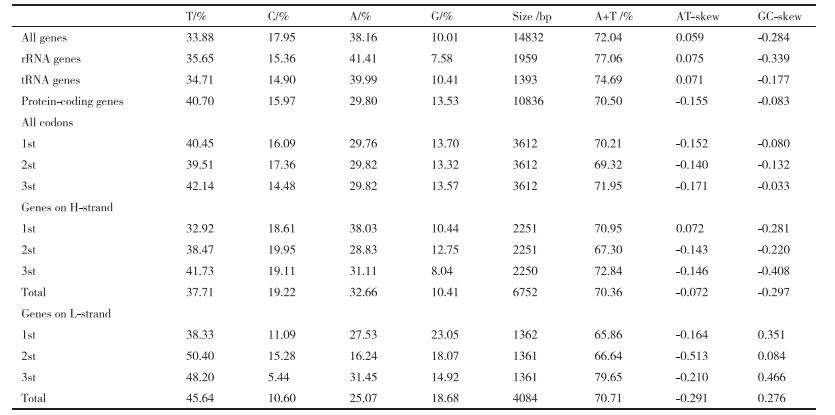
由表 3看出蛋白质编码基因密码子3个位置的碱基组成情况。3个位置的A+T含量均远高于G+C含量, 且密码子第3位的A+T含量最高, AT-skew绝对值为0.155, 而GC-skew绝对值仅为0.083。

使用Mega 6.0软件分析木通红喀木虱13个蛋白质编码基因的相对同义密码子使用频率(Relative synonymous codon usage, RSCU)。对于有两个同义密码子的氨基酸, 第3位点为A和U的密码子使用频率较高; 对于有4个同义密码子的氨基酸来说, H链编码基因偏向于使用第3位点为A的密码子, 而L链偏向于使用第3位点为U的密码子(表 3)。
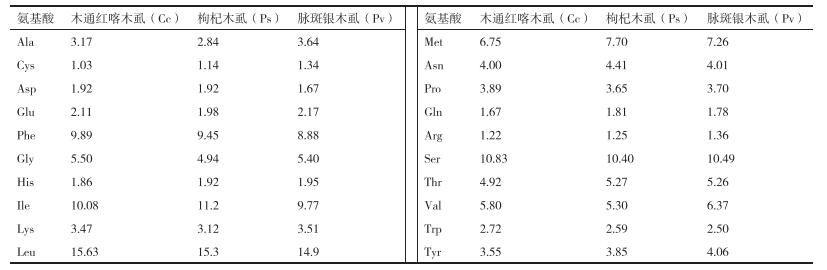
分析密码子使用情况可知, 木通红喀木虱线粒体中有6种密码子的使用率很高, 由高到低依次为UUU(F)、UUA(L)、AUU(I)、UCU(S2)、UAU(Y)和AAU(N)。分析氨基酸组成发现, Leu氨基酸编码量最高为15.63%;其次Ser、Ile和Phe分别为10.83%、10.08%和9.89%。而Cys最低仅为1.03%(表 4)。

木通红喀木虱线粒体基因组上的两个核糖体RNA基因(rrnL和rrnS)长度分别为1 153 bp和806 bp, 两者分别位于trnL和trnV, trnV和CR之间, 即trnV将两个rRNA间隔开(图 2)。分析碱基组成可得rRNA基因A+T含量为77.06%, A+T含量偏向性明显, 且AT-skew值为0.075, GC-skew值为-0.339。

|
| 图 2 通红喀木虱线粒体基因组rRNA基因 A:16S rRNA; B:12S rRNA |
木通红喀木虱线粒体基因组上的22个tRNA基因总长为1 393 bp, 其中最长72 bp, 而最短54 bp, 仅有一个trnS1不能形成三叶草结构。多数tRNA形成典型的三叶草结构, 非典型三叶草结构的tRNA情况各异(图 3)。

|
| 图 3 通红喀木虱线粒体基因组rRNA基因 |
其中trnA、trnD、trnQ、trnS2、trnT、trnV的TΨC臂上存在严重错配碱基; trnA、trnE、trnF、trnE1氨基酸接受臂上各有一处严重错配; trnD、trnG、trnT的TΨC臂缺失; trnL1、trnS2、trnW的TΨC环缺失。D环和TΨC环为3-11 bp, 除trnD、trnE反密码子环为11 bp外, 其他反密码子环均为9 bp。反义密码子臂含4-5对碱基, 而TΨC臂、D臂含2-4对碱基。木通红喀木虱线粒体基因组的22个tRNA上共有37处碱基错配。其中氨基酸接受臂上有14处错配, D臂上有9处错配, TΨC臂上有8处错配, 反密码子臂上有6处。
2.2 基因重叠和间隔序列将木通红喀木虱线粒体基因组的基因间隔区和重叠区与木虱科的其他两种木虱进行比较(表 5)。木通红喀木虱线粒体基因组的间隔区最短为106 bp; 脉斑银木虱线粒体基因组的间隔区最长为164 bp。而脉斑银木虱线粒体基因组的重叠区最短为45 bp; 枸杞木虱线粒体基因组的重叠区最长为143 bp。

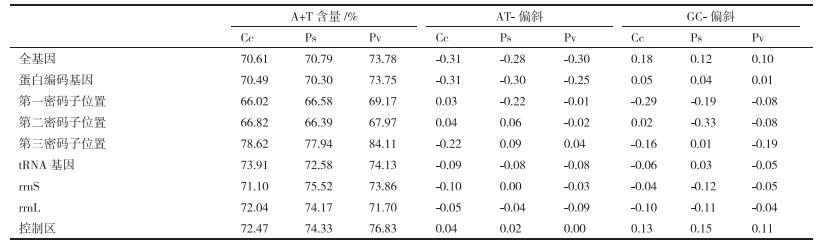
3种木虱线粒体基因组的碱基组成基本一致, 木通红喀木虱与枸杞木虱线粒体全基因的AT含量极接近均为70%左右, 而脉斑银木虱线粒体全基因的AT含量较多为73.78(表 6)。3种木虱均有显著的AT偏向。就线粒体基因而言, 木通红喀木虱的AT-Skew值为-0.31, GC-Skew值为0.18;枸杞木虱的AT-Skew值为-0.28, GC-Skew值为0.12;脉斑银木虱的AT-Skew值为-0.30, GC-Skew值为0.10。
由表 6可知3种木虱线粒体基因组中非编码区均位于rrnS和trnI之间, 木通红喀木虱、枸杞木虱和脉斑银木虱非编码区的长度分别为671 bp、744 bp和596 bp。
2.5 蛋白编码基因 2.5.1 蛋白编码基因的碱基组成木通红喀木虱、枸杞木虱和脉斑银木虱3种木虱线粒体基因组中, 蛋白质编码基因密码子第1、2和3位点在J-链和N-链上所使用碱基的情况如图 4。

|
| 图 4 三种木虱线粒体基因组碱基组成 |
如表 7, 三种木虱线粒体基因组中蛋白质氨基酸含量较高的前6位由高到低大致为亮氨酸(Leu)、丝氨酸(Ser)、异亮氨酸(Ile)、苯丙氨酸(Phe)、甲硫氨酸(Met)和缬氨酸(Val)此6种疏水性氨基酸。通过计算得出, 在木通红喀木虱食木虱、枸杞木虱和脉斑银木虱中6种疏水氨基酸的含量之和分别达到59.14%、59.03%和58.95%。
由图 5可知, 13个蛋白编码基因之间的ω值差异显著。

|
| 图 5 三种木虱线粒体基因组中蛋白质编码基因的序列分化比较 |
木通红喀木虱线粒体基因组共有14处重叠, 总长度为100 bp, 最大的重叠区域位于nad4和nad4L之间, 长度为49 bp, 最小的重叠区域有4处, 均为1 bp。而基因间隔区数量为11处, 总长度为106 bp, 最大的间隔仅为28 bp, 最小的间隔为1 bp。木通红喀木虱全线粒体基因组的碱基偏向性十分明显, 其中碱基A的含量最高为38.16%, 碱基T、C、G的含量分别为33.88%、17.95%和10.01%。蛋白质编码基因整体的A+T含量高于G+C含量。这一结果符合昆虫线粒体基因组的碱基组成上的A-T偏向性。蛋白编码基因的G、C碱基含量偏斜度低于A、T碱基, 这与线粒体基因组整体的分析结果相反, 蛋白质编码基因中碱基A与碱基T的含量上差值大于碱基G和碱基C含量差值。
木通红喀木虱的蛋白质编码基因以典型的ATN作为起始密码子。ATA是最为常用的起始密码子, cox2、ATP8、cox3、nad3、nad4L、nad6、cob和nad1这8种蛋白编码基因使用ATA作为起始密码子。而以ATT作为起始密码子的有2个nad5和nad2, 其余3个CoxI、ATP6和nad4使用ATA为起始密码子。13个蛋白质基因中仅有2个基因未在基因3'端找到完全的终止密码子。cox1、ATP8、ATP6、cox3、nad6、nad2此6个基因的终止密码子为TAA。其余5个基因nad3、nad4、nad4L、Cob和nad1以TAG为终止密码子。2个基因CoxII和nad5没有找到完整的终止密码子, 以T作为终止密码子。而造成这种原因可能是该位置与其后的tRNA基因重叠, 公用了几个碱基。除这2个不完整地终止密码子外, 木通红喀木虱的蛋白质编码基因共使用了3 530个密码子。
三种木虱线粒体基因组的碱基组成基本一致, 三者的AT偏斜均大于CG偏斜, 且偏斜明显。三种木虱的蛋白编码基因密码子第3位点的AT含量均明显高于其他2个位点。
木通红喀木虱、枸杞木虱和脉斑银木虱三种木虱线粒体基因组中, 分析可知三种木虱线粒体基因组上碱基组成基本相似, 其中J-链上各碱基含量与N-链的各碱基含量大致相同。且除枸杞木虱N-链上第2位密码子和脉斑银木虱N-链上第3位密码子外, 其余所有密码子位点的T含量均高于其它3种碱基的含量。三种木虱N链和J链上所有的密码子位点的AT含量均高于GC含量。
根据3种木虱线粒体基因组中蛋白质编码基因的序列所推导得出的氨基酸组成情况可知其含量大致相同, 且疏水氨基酸的大量出现, 符合线粒体基因编码的蛋白质多为跨膜蛋白这一特性。由蛋白编码基因之间的ω值差异显著表明不同的基因所承受的选择性压力不同(若非同义/同义置换率的比率(ω=Ka/Ks)大于1, 则说明蛋白质编码基因受到正选择, 若小于1, 则说明其受到负选择)。三种木虱mtDNA中J链上除nd6和cob基因外所有蛋白质编码基因的ω值大于1, 表明这些基因在进化过程中受到正选择。N链上除nad4L外所有基因的ω值均小于1(nad4L基因的置换速率是其它值的3倍左右), 说明N链大多基因受到负选择作用。
4 结论(1) 木通红喀木虱全线粒体基因组长度14 832 bp(NCBI登录号为NC_027087.1), A+T含量为72.04%。此线粒体基因组共编码37个基因, 包括13个蛋白质编码基因、22个转运RNA基因, 2个核糖体RNA基因, 这些基因的位置以及结构都很保守。
(2) 木通红喀木虱的蛋白质编码基因以典型的ATN作为起始密码子。ATA是最为常用的起始密码子, 此外还有ATT和ATA作为起始密码子。6个基因的终止密码子为TAA。5个基因以TAG为终止密码子。2个基因没有找到完整的终止密码子, 以T作为终止密码子。木通红喀木虱线粒体中使用率最高的6种密码子依次为UUU(F)、UUA(L)、AUU(I)、UCU(S2)、UAU(Y)和AAU(N)。分析氨基酸组成发现, Leu氨基酸编码量远高于其他, 为15.63%。其次Ser、Ile和Phe也很高, 分别为10.83%、10.08%和9.89%。而Cys最低, 仅为1.03%。
(3) 木通红喀木虱线粒体基因组上的两个核糖体RNA基因(rrnL和rrnS)长度分别为1 153 bp和806 bp。木通红喀木虱线粒体基因组上的22个tRNA基因仅有一个trnS1不能形成三叶草结构。
(4) 木虱科3种木虱枸杞木虱、木通红喀木虱、脉斑银木虱, 37个基因以及非编码区的排列顺序完全相同, 且三种木虱线粒体基因组的碱基组成基本一致, 同时氨基酸的组成情况及含量也大致相同。
| [1] |
Weintraub PG, Beanland LA. Insect vectors of phytoplasmas[J]. Annu Rev Entomol, 2006, 51: 91-111. DOI:10.1146/annurev.ento.51.110104.151039 |
| [2] |
张晓蓉, 陈义光. 湘西地区木通属植物调查[J]. 吉首大学学报:自然科学版, 2001, 22(2): 38-40. |
| [3] |
Martinez AL, Wallace JM. Citrus leaf mottle-yellows disease in the Philippines and transmission of the causal virus by a psyllid, Diaphorina citri[J]. Plant Dis Rep, 1967, 51(8): 692-695. |
| [4] |
Yang XB, Zhang YM, Hua L, et al. Life history and life tables of Bactericera cockerelli(Hemiptera:Psyllidae)on potato under laboratory and field conditions in the lower Rio Grande Valley of Texas[J]. J Econ Entomol, 2010, 103(5): 1729-1734. DOI:10.1603/EC10083 |
| [5] |
Hodkinson ID. The biology of the Psylloidea(Homoptera):a review[J]. Bulletin of Entomological Research, 1974, 64(2): 325-338. DOI:10.1017/S0007485300031217 |
| [6] |
Tsai JH, Liu YH. Biology of Diaphorina citri(Homoptera:Psyllidae)on four host plants[J]. J Econ Entomol, 2000, 93(6): 1721-1725. DOI:10.1603/0022-0493-93.6.1721 |
| [7] |
Liu D, Trumble J T. Comparative fitness of invasive and native populations of the potato psyllid(Bactericera cockerelli)[J]. Entomologia Experimentalis et Applicata, 2007, 123(1): 35-42. DOI:10.1111/eea.2007.123.issue-1 |
| [8] |
Sun J R, Li Y, Yan S, et al. Microsatellite marker analysis of genetic diversity of Cacopsylla chinensis(Yang et Li)(Hemiptera:Psyllidae)populations in China[J]. Acta Entomol Sin, 2011, 54: 820-827. |
| [9] |
陈慈相, 张倩, 谢金招, 等. 赣南地区柑桔黄龙病发生规律研究[J]. 中国南方果树, 2015, 44(6): 43-45. |
| [10] |
黄建昌, 梁关生, 刘小冬, 等. 几种杀虫剂对柑橘木虱的防治试验初报[J]. 中国园艺文摘, 2018, 1: 47-48. DOI:10.3969/j.issn.1672-0873.2018.05.019 |
| [11] |
唐毅.西南地区木虱总科区系分类研究(半翅目: 胸喙亚目)[D].贵阳: 贵州大学, 2007.
|
| [12] |
魏书军, 陈学新. 昆虫比较线粒体基因组学研究进展[J]. 应用昆虫学报, 2012, 48(6): 1573-1585. |
| [13] |
于卫卫.螺旋粉虱种群分化及线粒体全基因组和核糖体基因序列分析[D].扬州: 扬州大学, 2012.
|
| [14] |
李天杰, 曹延祥, 赵红翠, 等. 动物线粒体基因组测序方法的研究进展[J]. 天津医药, 2016(6): 796-800. |
| [15] |
Saccone C, Pesole G. Handbook of comparative genomics:principles and methodology[J]. Handbook of Comparative Genomics Principles & Methodology, 2003(4): 366-368. |
| [16] |
Thao MLL, Baumann L, Baumann P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids(Hemiptera, Sternorrhyncha)[J]. BMC Evolutionary Biology, 2004, 4(30): 25-25. |
| [17] |
Nishibori M, Tsudzuki M, Hayashi T, et al. Complete nucleotide sequence of the Coturnix chinensis(Blue-breasted quail)mitochondorial genome and a phylogenetic analysis with related species[J]. Journal of Heredity, 2002, 93(6): 439-444. DOI:10.1093/jhered/93.6.439 |
| [18] |
Yamauchi MM, Miya MU, Nishida M. Complete mitochondrial DNA sequence of the swimming crab, Portunus trituberculatus(Crustacea:Decapoda:Brachyura)[J]. Gene, 2003, 311: 129-135. DOI:10.1016/S0378-1119(03)00582-1 |
| [19] |
Simon C, Frati F, Beckenbach A, et al. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers[J]. Ann Entomol Soc Am, 1994, 87(6): 651-701. DOI:10.1093/aesa/87.6.651 |
| [20] |
Saccone C, Pesole G. Handbook of comparative genomics:principles and methodology[J]. Handbook of Comparative Genomics Principles & Methodology, 2003(4): 366-368. |
| [21] |
Thao MLL, Baumann L, Baumann P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids(Hemiptera, Sternorrhyncha)[J]. BMC Evolutionary Biology, 2004, 4(30): 25-25. |
| [22] |
Yamauchi MM, Miya MU, Nishida M. Complete mitochondrial DNA sequence of the swimming crab, Portunus trituberculatus(Crustacea:Decapoda:Brachyura)[J]. Gene, 2003, 311: 129-135. DOI:10.1016/S0378-1119(03)00582-1 |
| [23] |
Zhang DX, Hewitt GM. Assessment of the universality and utility of a set of conserved mitochondrial cox1 primers in insects[J]. Insect Molecular Biology, 1997, 6(2): 143-150. DOI:10.1111/imb.1997.6.issue-2 |