




2. 黑龙江东方学院食品与环境工程学部,哈尔滨 150066
2. Department of Food and Environment Engineering, East University of Heilongjiang, Harbin 150066
大豆作为一种重要的油料作物,在国民经济中一直占有重要地位。近年来,随着人们需求碳量的增大,耕地面积紧张导致大豆连作现象日益严重。黑龙江省是我国非转基因大豆主要产区,然而,大豆连作现象极为严重[1]。作物连作会造成土壤理化性质恶化、土传病害加剧及自毒作用,进而减少作物产量,影响作物正常生产[2-3]。在农田中,连作条件下大多依靠施肥来提高作物产量,但会破坏土壤结构、养分失衡、自毒物质积累,加剧作物土传病害的产生[4]。
细菌群落结构在土壤养分中一直扮演重要角色[5]。研究表明,连作会破坏土壤微生物群落结构,促进病原微生物生长,抑制有益微生物繁殖,进而造成作物减产[6-7]。相关研究证实,连作障碍可危害黄瓜,番茄和大豆等大多数作物的生长[8-10]。研究发现,土壤受土壤种类、种植模式和种植作物种类等众多因素影响,与间作种植模式相比,连作会导致土壤酶活性、微生物群落数和土壤养分均有降低[11]。近年来,有关根际土壤微生物菌群结构的研究多采用DGGE技术,但该技术仅能分析有限的优势微生物类群,存在高估物种丰度以及低估微生物群落大小和多样性的可能,并具有较大的随机性很难检测出低丰度的土壤微生物类群[12-13]。随着测序技术的发展,高通量测序已成为定量和鉴别微生物群落演替的更好的研究工具[14]。该技术能够在整体微生物细菌群落水平下分析物种遗传多样性,并能较为客观的反映样本中低丰度的重要功能微生物[15]。该技术具有测序通量高、试验过程简化、速度快、准确率高等优点,试验结果能够更好地反应出群落结构的特点。
本研究选用大豆品种为黑农48,大田选取连作0年和连作2年土样。利用Illumina高通量测序技术分析对比根际土壤中的细菌多样性旨在探索连作大豆根际土壤细菌群落结构,旨为改善连作大豆土壤菌群结构、提高大豆生长奠定前期理论基础。
1 材料与方法 1.1 材料试验在哈尔滨工业大学糖业研究所试验站进行。试验土壤主要理化指标:有机质26.13 g/kg,全N 1.69 g/kg,全P 5.5 g/kg,全K 24.6 g/kg,碱解N 136.2 mg/kg,速效P 13.40 mg/kg,速效K 205 mg/kg,pH7.0。
试验材料由黑龙江省农业科学院提供。选用黑龙江省主栽大豆品种:黑农48(HN48,高蛋白品种,蛋白质含量平均45%,脂肪含量平均为20%)。
1.2 方法 1.2.1 大田试验设计试验土壤分别选用:连作0年(未种植大豆土壤,即第一年种植大豆土壤,三个生物学重复分别标记为L0_1、L0_2和L0_3)和连作2年(种植过2年大豆,及第三年种植大豆土壤,三个生物学重复分别标记为L2_1、L2_2和L2_3)大田土壤。2016年5月17日播种,生育期正常浇水。
1.2.2 样本采集在大豆成熟期进行采样,三点取样法随机取样,取10-20 cm大豆根际土壤样本,每个土样随机采取三点,去除土样中的植物和动物残体等杂质,分别混合后过40目筛子,一部分装入土袋中保存于-80℃冰箱中; 另一部分用于土样的高通量测序。
1.2.3 Illumina高通量测序土壤样本用干冰送到上海美吉生物医药科技有限公司,进行Illumina高通量测序。细菌的16S rDNA扩增引物选用V3-V4区引物(338F-806R),每个土壤样本3个生物学重复。
1.2.4 有效序列和优化序列数据统计运用多个样品平行测序的方法,所以各样品中的序列均引入了一段标示其样本来源信息的barcode标签序列及前引物(forward primer)序列。本次分析根据barcode标签序列和前引物序列筛选出有效序列后,将测序接头与barcode序列去除,并对处理后的有效序列进行数据统计。为了得到更高质量及更精准的生物信息分析结果,应对有效序列进行去杂。丢弃长度短于150 bp、含有模糊碱基、引物碱基含2个以上错配的序列,即得到优化序列,之后对优化序列进行数据统计。
1.2.5 OTU-based分析对所有样品进行OTU生成并对OTU进行生物信息统计分析。将优化序列选取长度大于350 bp的序列并截齐后与silva库比对后对序列进行聚类。聚类分析使用的软件为mothur和chopseq。
1.2.6 细菌群落多样性分析细菌群落物种的丰富度和多样性分别用Chao指数和Shannon指数及Simpson指数表示,测序深度指数用Coverage表示。
1.2.7 群落结构分析将优化序列根据silva库中的参考序列鉴定OTU种属。在门的水平上做样品群落分布柱状图,比较3个土样中的菌落分布情况。
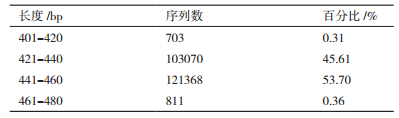
2 结果 2.1 测序数据分析通过Illumina高通量测序并优化后,样品共获得225 998条序列,总碱基数为136 050 796 bp,平均碱基长度为99 228 256 bp,其中421-440 bp和441-460 bp的碱基分别占总序列的45.61%和53.70%,如表所示。
对测序获得的序列进行随机抽样,以抽样得到的序列数与他们所代表的OTU数目构建稀释性曲线,从图 1中可以看出,6个样本的稀释性曲线菌区域平坦,这表明试验测序数据合理,更多的测序数据只会产生少量新的OTU。

|
| 图 1 稀释曲线图 |
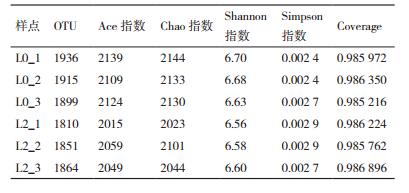
通过比较DNA序列的相似性,将大于97%的序列相似性归为同一种可操作分类单元(opertinal taxonomic unit,OTU)。对不同连作年限的大豆根际土壤细菌16S rDNA多样性指数进行分析,L0_1、L0_2、L0_3、L2_1、L2_2及L2_3的Shannon指数分别为6.70、6.68、6.63、6.56、6.58及6.60,Chao指数分别为2 144、2 133、2 130、2 023、2 101和2 044(表 2),可以看出连作后细菌的多样性和丰富度均有所降低。表明不同连作年限的大豆根际土壤的细菌群落结构存在差异,细菌多样性及丰富度均降低。
通过表 3的独立样本检验发现方差方程的Levene检验P=0.294>0.05证明具有方差齐性; 则P=0.019<0.05证明连作0年与连作2年间的香农指数存在差异显著。

由图 2可知,全部供试土壤中共检测到10个细菌门,其中L2年中相对丰度大于1%的有10个,L0年的有9个。80%以上的细菌序列属于变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)。而牙单胞菌门(Gemmatimonadetes)、拟杆菌门(Bacteroidetes)、疣微菌门(Verrucomicrobia)、硝化螺菌门(Nitrospirae)、厚壁菌门(Firmicutes)、浮霉菌门(Planctomycetes)的相对丰度所占比例较低。

|
| 图 2 连作2年与连作0年门水平群落结构组成图 |
随着连作年限的增加细菌的门水平也随之产生变化,变形菌门(Proteobacteria)的相对丰度连作0年较连作2年有所增加,相似的放线菌门(Actinobacteria)也出现了丰度增加现象。而相反的酸杆菌门(Acidobacteria)和绿弯菌门(Chloroflexi)的相对丰度在连作2年要低于连作0年。
同时从数据库中鉴定出418个属,从土壤样本细菌属的相对丰度中能发现年际变化。从图 3可以看出大豆连作0年最丰富的细菌属是Subgroup_6_norank,Anaerolineaceae和RB41_norank,而连作2年的细菌优势属为Subgroup_6_norank,Acidimicrobia-les,Gemmatimonadaceae,Nitrosomonadaceae和Ana-erolineaceae。

|
| 图 3 连作2年与连作0年属水平群落结构组成图 |
由图 3可知,通过对比分析发现连作大豆2年土样中的酸微菌目(Acidimicrobiales_uncultured)、硝化螺菌属(Nitrospira)、鞘氨醇菌属(Sphingomo-nas)和芽孢杆菌属(Bacillus)的相对丰度要高于连作0年土样。
2.3 群落结构分析在属的水平上对样品和样品所含菌属进行聚类,根据聚类后的各样品中不同OTU数,对应所含序列的丰度作出Heatmap(图 4),颜色梯度的变化,能够反映出在菌属水平上各样品细菌群落结构的差异性。菌属的丰度会受到大豆连作条件的影响,其中菌属的丰度受作物种植方式影响最大的是Acinobacteria、Proteobacteria、Acidobacteria、Chlor-oflexi和Verrucomicrobia。在连作与轮作土壤中所测得的细菌菌属中丰度差异较大的有:Acidimicrobia-les、Gemmationadaceae、Nitrosomonadaceae和Anare-olinenceae等。

|
| 图 4 基于16S rDNA序列构建的热图 |
由图 5可见,可以将大豆根际土壤16S rDNA细菌的Heatmap图划分为3个Cluster。首先Cluster1主要包括亚硝化细菌科、酸微菌目、厌氧绳菌科。大豆种植方式的改变影响着大豆根际土壤中的细菌相对丰度。连作2年中的酸微菌目丰度增加高于连作0年。Cluster2主要包括红色杆菌属、假诺卡氏菌属、噬纤维素菌科以及一些未知的菌,其中红色杆菌属与噬纤维菌科相对丰度有所减少,而假诺卡氏菌属略有增加。Cluster3中主要包括牙单胞菌属、链霉菌属、慢生根瘤菌属等。从图 4可见,芽单胞菌属在连作2年种植后丰度降低,但是链霉菌属、及慢生根瘤菌属在连作种植后细菌丰度高于轮作种植。

|
| 图 5 基于16S rDNA序列构建的热图 |
从总体来看,连作2年与连作0年相比,细菌群落结构和丰度变化并不显著。这说明连作种植模式虽然会降低根际土壤中细菌多样性但对细菌的群落结构和丰度影响较小。
3 讨论从影响部位看,连作造成的微生物群落变化主要发生在根际部位。虽然可培养微生物占土壤微生物总量的比例不足1%,但土壤中可培养细菌是对土壤生态系统贡献最大的类群,它们比整个微生物群体更容易遭受土壤生态系统变化的影响,可用作与污染影响有关的指示剂[16-17]。土壤微生物几乎参与土壤中一切生物和生物化学反应,其主要生理类群则直接参与土壤中C、N等营养元素的循环和能量流动,其数量和活性直接关系到土壤生态系统的维持和改善[18]。由此连作根际土壤中菌群结构变化一直是人们研究过程中的重点。
本实验应用高通量测序技术得到的各处理Coverage指数均达到80%以上,证实本次测序结果可以反应样本的真实情况。对于分析根际土壤中的不同微生物群落结构是十分有效的方法,多样性指数数值越高则代表样本中的微生物群落结构多样性越高,它由群落的多样性及群落的丰富度两部分构成[19-20]。本研究结果表明,连作2年较连作0年土壤细菌的群落结构有明显差异,Shannon指数和丰富度指数均呈现L0>L2。可见随着连作种植模式的出现,大豆根际土壤中的细菌多样性随之减少。大量研究证实连作会破坏土壤细菌菌群结构,降低土壤细菌多样性及菌种的丰富度[3, 21]。
本实验研究中,连作年限不同的根际土壤中细菌群落结构存在一定的差异。连作0年土样中的细菌Shannon指数要高于连作2年土样且差异显著,这一结论表明连作后的土壤多样性指数有所下降。同时通过试验结果得知放线菌门(Actinobacteria)在连作2年与连作0年中表现出增加趋势。连作种植后土样中的放线菌数量增加,这一结果与孙秀山等[22]的研究结果相似。揭示长期连作会对细菌,真菌和放线菌造成的影响较大,而连作障碍在短时期内对细菌与放线菌造成的影响较小[22]。大蒜在连作种植后,土壤中的细菌与放线菌数量均随种植年限的增加而表现出先上升后下降态势,在连作种植某年为期,在此之前细菌和放线菌的数量及微生物总量逐渐增多,之后猛然下降[23]。另外一些研究结果也有显示,连作并没有使土壤由细菌型向真菌型转变。例如对地黄、太子参的相关研究证实,随着连作年限的增加,土壤细菌数量下降极为显著,但放线菌数量却增加极为显著[24-25]。
通过Heatmap图和群落结构图得知,大豆连作种植后根际土样中的酸微菌目、根瘤菌属、芽孢杆菌属及鞘氨醇菌属的相对丰度均大于轮作土样,而纤维分解菌属在连作土样中相对丰度下降。且得出的试验结果在其他作物种植过程中也有出现,如固氮菌与纤维素分解菌都作为有益微生物分别参与了土壤的氮素与碳素循环过程。李琼芳[26]研究证实随连作年限的增加,土壤中有益细菌数量锐减; 当大蒜连作5-10年后,土壤中的硝化细菌和鞘氨醇单胞菌数量呈上升趋势,并未有连作障碍的现象出现,只有经过连作15-20年后才会出现细菌数量的减少等明显连作障碍现象[27]。李婧等[28]通过克隆文库的研究方法证实,花生在连作后,土壤中的芽孢杆菌的群落结构发生改变,其中的一些芽孢杆菌如高地芽孢杆菌成为了土壤微生态环境的优势菌群。同时在不同作物连作后,土壤细菌生理菌群的变化趋势不尽相同:刘亚峰等[29]对黄瓜连作表明连作2年土壤中自生固氮菌和纤维素分解菌数量均上升,第3年有所下降。而陈慧等[24]对地黄连作的研究表明连作地黄土壤中氨化细菌、反硝化细菌、好气性固氮菌、嫌气性纤维素分解菌、硫化细菌数量增多,好气性纤维素分解菌大量减少。通过研究发现连作种植后,土壤中有益的芽孢杆菌、纤维素分解菌及固氮菌等有益细菌的数量增加,这与以上研究结果相一致。有益细菌未显著减少表明细菌菌群结构变化并不显著。
从Heatmap图和群落结构图的总体上看,细菌的多样性和丰度变化不大,这也与大多数田间观察结果相同。例如花生在连续种植期间根际细菌数量逐渐降低、真菌数量明显上升[30]。通过对连作五年马铃薯的观察发现其土壤中的尖孢镰刀菌和立枯丝核菌等数量增加,土传病害愈加严重,进而影响作物生长。作物病虫害更加严重。连作环境保持相对稳定的状况会导致病虫害的频发。连作对细菌以及固氮菌均有抑制作用,而对真菌却有着促进作用,土壤由细菌型转换为真菌型,由高肥力转变为低肥力[31, 32],连作后土壤中原有的优势属的线虫群落如食细菌线虫和食真菌线虫显著降低,而以植物寄生线虫为优势属的线虫开始破坏寄主植物,致使土壤不利于作物生长,进而造成减产[33]。由此可见致使作物连作的主要影响因素是真菌病害及线虫病害,而细菌数量会逐渐降低,真菌病害逐年严重,这也与Heatmap图细菌丰度变化较小的趋势吻合。由此推测,连作土壤中细菌并不能够占据主导地位,对连作的影响并不明显,相较真菌的优势性来看,细菌在土壤中发挥的作用较小。本研究结果表明大豆连作2年较连作0年硝化细菌和固氮菌数量均有减少的变化趋势。由此推测在短期连作种植过程中,不同作物根际微生物类群的变化规律各不相同,难以用来推断细菌在作物连作所存在普遍现象。
本研究应用高通量测序技术探究连作2年与连作0年土壤中的细菌群落结构变化。通过对两种大豆种植模式根际土壤分析可知,连作0年土壤的多样性指数高于连作2年土壤,这表明大豆的连作种植对土壤细菌群落结构有重要影响,但相对丰度较低且变化并不显著,证明在连作土壤中细菌并不能占据主导地位发挥作用。因此,也有必要研究长期连作过程细菌群落结构的组成及多样性随着连作年限的延长发生的变化。
4 结论研究结果与其他相关研究报道有相似之处,发现连作后土壤的有益细菌增加,如:鞘氨醇单胞菌、固氮菌、纤维素分解菌等。同时试验发现连作2年土样的微生物多样性要低于连作0年,推测短期大豆连作虽然使细菌菌群多样性降低,但是在大豆连作2年时并没有产生严重的连作障碍现象,土壤中细菌菌群丰度也并未发生显著变化。
| [1] |
李亮, 魏丽娜, 崔佳琦, 蔡柏岩. 摩西管柄囊霉Funneliformis mosseae对连作大豆分枝期根系AM真菌群落结构的影响[J]. 菌物学报, 2016, 35(7): 882-891. |
| [2] |
Liu X, Zhang J, Gu T, et al. Microbial community diversities and taxa abundances in soils along a seven-year gradient of potato monoculture using high throughput pyrosequencing approach[J]. PLoS One, 2014, 9(1): e86610. DOI:10.1371/journal.pone.0086610 |
| [3] |
She S, Niu J, Zhang C, et al. Significant relationship between soil bacterial community structure and incidence of bacterial wilt disease under continuous cropping system[J]. Archives of Microbiology, 2017(199): 267-275. |
| [4] |
Li X, Lewis EE, Liu Q, et al. Effects of long-term continuous cropping on soil nematode community and soil condition associated with replant problem in strawberry habitat[J]. Scientific Reports, 2016, 6: 30466. DOI:10.1038/srep30466 |
| [5] |
刘方春, 邢尚军, 马海林, 等. 持续干旱对樱桃根际土壤细菌数量及结构多样性影响[J]. 生态学报, 2014, 34(3): 642-649. |
| [6] |
Klein E, Katan J, Gamliel A. Soil suppressiveness to fusarium disease following organic amendments and solarization[J]. Plant Disease, 2011, 95(95): 1116-123. |
| [7] |
Dong L, Jiang X, Feng G, et al. Soil bacterial and fungal community dynamics in relation to Panax notoginseng death rate in a continuous cropping system[J]. Scientific Reports, 2016, 6: 31802. DOI:10.1038/srep31802 |
| [8] |
Lu L, Yin S, Liu X, et al. Fungal networks in yield-invigorating and -debilitating soils induced by prolonged potato monoculture[J]. Soil Biology and Biochemistry, 2013, 65: 186-194. DOI:10.1016/j.soilbio.2013.05.025 |
| [9] |
Li B, Cui J, Jie W, Cai B. Analysis of the community compositions of rhizosphere fungi in soybeans continuous cropping fields[J]. Microbiol Res, 2015, 180: 49-56. DOI:10.1016/j.micres.2015.07.007 |
| [10] |
Zhang XP, Ning TY, Yang Y, et al. Effects of different application rates of calcium cyanamide on soil microbial biomass and enzyme activity in cucumber continuous cropping[J]. Chinese Journal of Applied Ecology, 2015, 26(10): 3073-3082. |
| [11] |
Yang R, Mo Y, Liu C, et al. The effects of cattle manure and garlic rotation on soil under continuous cropping of watermelon(Citrullus lanatus L.)[J]. PLoS One, 2016, 11(6): e0156515. DOI:10.1371/journal.pone.0156515 |
| [12] |
Tan Y, Chen Q, Liu H J, et al. Bacteria community in different aged Coptis chinensis planting soil revealed by PCR-DGGE analysis[J]. China Journal of Chinese Materia Medica, 2015, 40(16): 3147. |
| [13] |
邢德峰, 任南琪. 应用DGGE研究微生物群落时的常见问题分析[J]. 微生物学报, 2006, 46(2): 331-5. |
| [14] |
Voříšková J, Baldrian P. Fungal community on decomposing leaf litter undergoes rapid successional changes[J]. Isme Journal, 2013, 7(3): 477-86. DOI:10.1038/ismej.2012.116 |
| [15] |
Cheng Y, Ma Q, Ren H, et al. Fine mapping of a Phytophthora-resistance gene RpsWY in soybean(Glycine max L.)by high-throughput genome-wide sequencing[J]. Theoretical & Applied Genetics, 2017, 130(5): 1041-1051. |
| [16] |
Ward DM, Weller R, Bbteson MM. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community[J]. Nature, 1990, 345(6270): 63-65. DOI:10.1038/345063a0 |
| [17] |
Ellis RJ, Morgan P, Weightman AJ, Fry JC. Cultivation-dependent and-independent approaches for determining bacterial diversity in heavy-metal-contaminated soil[J]. Applied & Environmental Microbiology, 2003, 69(6): 3223-3230. |
| [18] |
许光辉, 郑洪元, 张德生, 等. 长白山北坡自然保护区森林土壤微生物生态分布及其生化特性的研究[J]. 生态学报, 1984, 4(3): 9-25. |
| [19] |
Harcha BD, Corrella RL, Meechb W, et al. Using the Gini coefficient with BIOLOG substrate utilisation data to provide an alternative quantitative measure for comparing bacterial soil communities[J]. Journal of Microbiological Methods, 1997, 30(1): 91-101. DOI:10.1016/S0167-7012(97)00048-1 |
| [20] |
张喜, 王莉莉, 刘延惠, 等. 喀斯特天然林植物多样性指数和土壤理化指标的相关性[J]. 生态学报, 2016, 36(12): 3609-3620. |
| [21] |
Tan Y, Cui Y, Li H, et al. Diversity and composition of rhizospheric soil and root endogenous bacteria in Panax notoginseng during continuous cropping practices[J]. Journal of Basic Microbiology, 2017, 57(4). |
| [22] |
孙秀山, 封海胜, 万书波, 左学青. 连作花生田主要微生物类群与土壤酶活性变化及其交互作用[J]. 作物学报, 2001, 27(5): 617-621. |
| [23] |
刘素慧, 刘世琦, 张自坤, 等. 大蒜连作对其根际土壤微生物和酶活性的影响[J]. 中国农业科学, 2010, 43(5): 1000-1006. |
| [24] |
陈慧, 郝慧荣, 熊君, 等. 地黄连作对根际微生物区系及土壤酶活性的影响[J]. 应用生态学报, 2007, 18(12): 2755-2759. |
| [25] |
林茂兹, 王海斌, 林辉锋. 太子参连作对根际土壤微生物的影响[J]. 生态学杂志, 2012, 31(1): 106-111. |
| [26] |
李琼芳. 不同连作年限麦冬根际微生物区系动态研究[J]. 土壤通报, 2006, 37(3): 563-565. |
| [27] |
周宝利, 徐妍, 尹玉玲, 叶雪凌. 不同连作年限土壤对茄子土壤生物学活性的影响及其嫁接调节[J]. 生态学杂志, 2010, 29(2): 290-294. |
| [28] |
李婧, 陈广波, 张坤, 崔中利, 曹慧. 花生连作红壤芽孢杆菌的群落多样性及其生防效果研究[J]. 土壤, 2012, 44(5): 776-781. |
| [29] |
刘亚锋, 孙富林, 周毅, 贾新成. 黄瓜连作对土壤微生物区系的影响Ⅰ——基于可培养微生物种群的数量分析[J]. 中国蔬菜, 2006, 1(7): 4-7. |
| [30] |
Compant S, Duffy B, Nowak J, et al. Use of Plant growth-promoting bacteria for biocontrol of plant diseases:principles, mechanisms of action, and future prospects[J]. Applied & Environmental Microbiology, 2005, 71(9): 4951-499. |
| [31] |
Yang R, Mo Y, Liu C, et al. The Effects of Cattle manure and garlic rotation on soil under continuous cropping of watermelon(Citrullus lanatus L.)[J]. PLoS One, 2016, 11(6): e0156515. DOI:10.1371/journal.pone.0156515 |
| [32] |
Xiong W, Zhao Q, Xue C, et al. Comparison of fungal community in black pepper-vanilla and vanilla monoculture systems associated with vanilla fusarium wilt disease[J]. Frontiers in Microbiology, 2016, 7: 117. |
| [33] |
马媛媛, 李玉龙, 来航线, 等. 连作番茄根区病土对番茄生长及土壤线虫与微生物的影响[J]. 中国生态农业学报, 2017, 25(5): 730-739. |