




2. 中国科学院大学资源与环境学院,北京 100049
2. College of Resources and Environment, University of Chinese Academy of Sciences, Beijing 100049
氮循环是生物地球化学循环的重要组成部分,现代氮循环几乎全部依靠微生物介导的氧化还原反应完成(图 1),只有小部分依靠长期的地质循环[1-2]。微生物,包括细菌、古菌和真菌,在氮循环中起着不可替代的作用,它们之所以能够实现氮素转化是由于体内存在一系列氮代谢相关的酶。编码这些酶的活性亚基的基因可作为相应微生物的功能标记基因[3],通过分子检测技术鉴定这些功能基因是我们了解环境中氮循环过程的重要手段和方法。

|
| 图 1 氮循环的各个生态过程及相关的功能基因 箭头是不同的氮素形态的转化过程,箭头上的基因是催化该转化过程的相关酶的标记基因。固氮作用:固氮酶基因nifH;硝化作用:氨单加氧酶基因amoA,羟胺氧化还原酶基因hao,亚硝酸盐氧化还原酶基因nxrA;反硝化作用:硝酸盐还原酶基因narG,亚硝酸盐还原酶基因nirS和nirK,一氧化氮还原酶基因norB,氧化亚氮还原酶基因nosZ;厌氧氨氧化作用:N2H4合成酶基因hzsA,N2H4氧化还原酶hzo;氮同化还原作用:硝酸盐同化还原酶基因nasA、narB,亚硝酸盐同化还原酶基因nirA、nirB;氮异化还原作用(DNRA):硝酸盐异化还原酶基因napA,亚硝酸盐异化还原酶基因nrfA;氨化作用:尿素酶基因ureC。 |
对参与氮循环的功能微生物的研究已有上百年历史,编码特定功能酶的功能基因不断成功破译,极大地拓展了对氮循环功能微生物的认识。尤其是近十几年来宏基因组学技术的革新,促使氮循环相关研究出现了一系列令人瞩目的成果,例如氨氧化古菌和完全硝化菌的发现更是颠覆了人们百年来对硝化作用的固有认识。早在19世纪,Winogradsky(1890年)分离的一株氨氧化菌便证明了细菌在硝化作用中发挥着作用,由此氨氧化细菌(AOB)被认为是氨氧化过程的主要参与者。直到2004年基于宏基因组学的研究发现,海洋古菌的基因组中含有与细菌类似的编码氨单加氧酶的结构基因[4];而次年从西雅图水族馆海水中分离培养得到第一株氨氧化古菌(AOA)[5],至此彻底改变了学术界认为只有AOB进行氨氧化作用的传统认识[6-7]。而2015年完全硝化菌成为了又一颠覆性发现,Daims等[8]和van Kessel等[9]对全球广泛分布的硝化螺旋菌属(Nitrospira)进行宏基因组高通量测序,发现这种化能自养菌拥有能够编码完成硝化作用将NH4+转化为NO2-再转化为NO3-的两大步骤必需的所有酶,比常规的氨氧化微生物生长率更低而生长量更高。他们这一重大发现颠覆了一直以来认为硝化作用的两大步骤不能被同一个微生物催化完成的固有观念,完全硝化菌也因此成为了氮循环微生物的重要成员。在回顾氮循环研究发展的历程中,我们发现在各个重要节点,功能基因分子检测技术都起到了关键性作用。本文将简要介绍PCR扩增技术、DNA指纹图谱技术、稳定性同位素示踪技术、分子杂交技术、测序技术等分子检测技术,重点论述其在氮循环功能微生物的群落多样性方面的研究进展,最后指出分子检测技术的革新和完善的数据分析平台的建立对未来氮循环功能微生物研究的重要意义。
1 分子检测技术的发展准确测量微生物丰度是开展微生物生态学研究的基础,但精确度量是一项具有挑战的任务,而分子技术的革新使得更为精准的度量成为可能[10]。宏基因组分子检测技术使用非培养的手段,通过环境样品中所有微生物的DNA来获得微生物群落遗传信息,能够更加全面、真实、精确地反映微生物群落的结构及潜在功能[11-12]。常用的分子检测技术分为五大类:PCR扩增技术、DNA指纹图谱技术、稳定性同位素示踪技术、分子杂交技术、测序技术(表 1)。
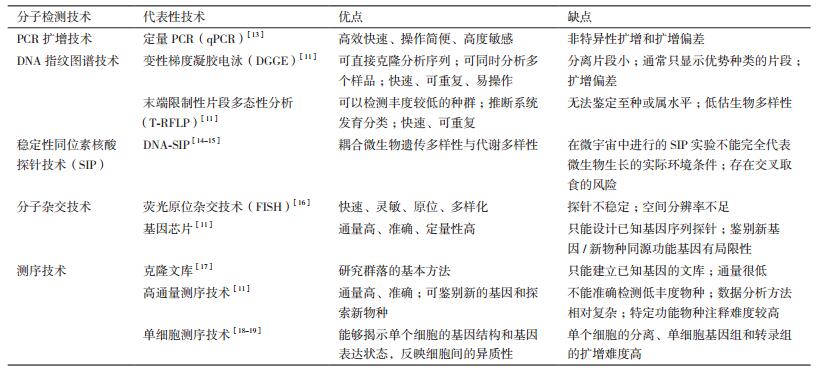
常用的分子检测技术中,基因芯片和高通量测序技术属于高通量分子检测技术,其他的技术通量较低。基因芯片技术通过一组已知核酸探针与环境样品杂交进行核酸序列测定[11]。基因芯片包括系统发育芯片(PhyloChip)和功能基因芯片(GeoChip)[20],系统发育芯片通过检测特异性的16S rRNA等系统发育标记基因,反映不同物种在特定环境的分布情况;功能基因芯片通过检测执行特殊功能的功能基因,来反映在微生物群落中的功能类群的分布情况。高通量测序技术分为扩增子测序和宏基因组鸟枪测序。扩增子测序是针对目标基因,如16S rRNA基因展开测序,包括焦磷酸测序,Miseq和Hiseq等;鸟枪测序不针对特定目标基因,随机地对环境样品基因组展开测序[11]。
此外,单细胞测序技术首先利用单细胞分离技术将细胞分离成个体,然后针对单个细胞的基因组进行高通量测序分析,能够揭示单个细胞的基因结构和基因表达状态,获取未培养微生物遗传信息,在发掘环境“暗物质”方面有很好的应用前景[18-19]。细胞内融合PCR技术(Emulsion paired isolation and concatenation PCR,epicPCR)[21]是2016年发表的技术,该技术的优势是可在未培养的单细胞中将功能基因与系统发育标记基因(如16S rRNA基因)连接在一起,通量可达数十万个细胞,而成本只与一个基因组文库相当。
自1991年克隆文库[22]应用于环境样本研究开始,FISH[23]、DGGE[24]、T-RFLP[25]、DNA-SIP[26]、qPCR[27]、基因芯片[28]、高通量测序[29-30]、单细胞测序[31-32]、epicPCR[21]这些分子检测技术也相继应用于环境样本研究(图 2-A)。最早应用于环境氮循环功能微生物研究的分子检测技术是克隆文库[33],始于1997年,随后FISH[34]、T-RFLP[35]、DGGE[36]、基因芯片[28]、qPCR[37]、DNA-SIP[38]、高通量测序[39]也相继应用于环境样本中氮循环功能基因的研究(图 2-B)。分子检测技术在帮助研究者有效识别氮循环各过程中的关键微生物类群、揭示不同环境中氮循环功能微生物与环境的互作机制方面起到了重要作用,为我们更深入地解析不同生态系统中氮循环过程对全球气候环境变化的响应与反馈奠定了理论基础。

|
| 图 2 不同分子检测技术应用于环境样本(A)和氮循环功能基因(B)的发展史 同种颜色的方框表示同一类的分子检测技术 |
固氮作用(Nitrogen fixation)是N2被还原成NH4+和其他含氮化合物的过程(图 1),微生物在此过程中发挥重要作用。有研究表明生物固氮作用贡献了生物圈中近一半的氮元素年输入总量[40],是自然生态系统和农业生态系统中重要的氮来源过程。固氮酶由nifD和nifK基因编码的异源四聚体以及nifH基因编码的二氮酶还原酶亚基两个组分构成[40]。nifH基因作为固氮作用的标记基因[41],主要基于两点考虑:一是,目前已知的所有固氮生物都包含nifH基因;二是,基于nifH基因的系统发育关系和16S rRNA基因的系统发育关系非常地接近[42]。nifH基因的同系物可分成5个主要的系统发育群系[43],而根据同源性收集的nifH基因数据库(http://www.css.cornell.edu/faculty/buckley/nifh.htm)可用于系统发育和进化的相关分析、探针/引物的设计和评估、以及固氮酶基因多样性的检测[40-42]。
基于nifH基因的高通量分子检测技术能更深入、全面、细致地揭示不同土壤生态系统中固氮微生物的组成、多样性及功能。在自然土壤固氮微生物的研究中,Soni等[42]使用基于PCR扩增的克隆文库方法研究了西印度喜马拉雅山脉土壤中的固氮微生物,结果表明大多数固氮微生物属于γ-变形菌(γ-Proteobacteria),且不同的土壤类型中的固氮菌群落结构不同。Wang等[44]应用454焦磷酸测序法研究腾格里沙漠植被恢复地区中100年来固氮微生物的演化,发现在植被恢复区表层土中进行固氮作用的微生物主要属于蓝藻(Cyanobacteria),蓝藻不仅维持着沙漠表土中的固氮收支平衡,对沙漠中的初级生产力也有极大的贡献。在农业土壤固氮微生物研究中,Wang等[45]使用qPCR和T-RFLP技术,分析了小麦生长季节中在四种不同施肥模式下、不同深度土壤中固氮微生物群落丰度和结构的变化;结果表明随着土壤深度的增加nifH基因和16S rRNA基因丰度下降,而土壤深度增加带来的土壤理化性质的变化是影响固氮微生物群落结构的主要因子,而非取样季节或施肥模式。Zhang等[46]使用ITPGM(Ion Torrent Personal Genome Machine)和454焦磷酸测序对四种不同作物类型和轮作方式的种植系统土壤样品中微生物的nifH基因进行检测,发现在5%氨基酸序列不相似性水平下,总共检测到6182个操作分类单元(Operational taxonomic units, OTU),现有作物类型和轮作历史是显著影响固氮微生物群落结构的重要因素。土壤生态系统的固氮微生物研究发现,自然土壤生态系统中固氮菌主要包括γ-变形菌和蓝藻,固氮菌对土壤植被恢复有一定的贡献,农业土壤生态系统中固氮菌受到土壤深度、作物类型和轮作历史带来的土壤理化性质的变化的显著影响。
在水体环境中,高通量分子检测技术也有更好的表现。Wang等[47]综合使用nifH基因克隆文库、DGGE法、qPCR、逆转录PCR法,研究发现蓝藻是中国东圳水库中主要的固氮微生物;水分层/混合状态和氮含量会导致固氮菌群落分布的垂直模式和季节模式的变化,从而影响水体的氮循环。Xiao等[48]使用焦磷酸测序法对中国南海从珠江三角洲到开阔海域200m以上的表层海水中的固氮菌群进行研究,发现固氮菌的多样性很低,主要由γ-变形菌和束毛藻属(Trichodesmium)组成,而水深是影响固氮微生物丰度和多样性的最重要的因素。综合土壤环境和水体环境中固氮菌的研究发现,γ-变形菌和蓝藻确实是占主导的固氮微生物。在水体环境中,水深是影响固氮菌垂直分布的主要原因。
454焦磷酸测序、qPCR、克隆文库等方法能较为全面地检测nifH基因在土壤环境和水体环境中的分布。其中,454焦磷酸测序法和qPCR的到的数据更为细致。但每一种技术都有其局限性(表 1),新的技术在解决之前技术局限性的同时,也会产生新的问题。在有完善的数据库的前提下,使用更新的高通量分子检测技术能更深入、全面、细致地揭示不同生态系统中固氮微生物的组成、多样性及功能。
3 硝化作用功能基因硝化作用(Nitrification)广泛存在于各种生态系统中,对生态系统生产力和营养物质循环以及废物处理都起着极为重要的作用[6]。该过程包含3个小步骤,NH4+氧化为NH2OH,进而被氧化为NO2-,最后NO2-被氧化为NO3-(图 1),对应的功能基因分别是amoA、hao[49]和nxrA基因[50]。原来认为氨氧化过程和亚硝酸盐氧化过程是由不同的微生物完成,而最新发现的完全硝化菌可以独自完成这两个过程[8-9]。
3.1 氨单加氧酶基因amoA氨单加氧酶(Amo)催化NH4+氧化为NH2OH,是氨氧化作用中重要的酶[51],Amo操纵子由amoA、amoB和amoC这3个结构基因构成,amoA基因编码Amo的活性蛋白亚基,且具有一定的序列保守性[49]。已经证实amoA基因和16S rRNA在系统发育上是一致的,且存在于所有氨氧化微生物中[52],因此被选作Amo酶的功能标记基因,常用于氨氧化古菌(AOA)和氨氧化细菌(AOB)的检测分析。通过amoA基因的分子检测,发现AOA和AOB广泛分布于土壤、污水处理系统和海洋环境中。
在农业土壤系统中,Cubillos等[53]使用基于16S rRNA和amoA基因的DGGE法研究了哥伦比亚不同畜牧系统对AOB群落的影响,发现传统单一牧场(Conventional monoculture pastures)中的AOB的丰度以及硝化潜力显著高于其他两个系统,多冠层密集型森林畜牧系统(Multi-canopy intensive silvopastoral systems,ISS)中的丰度相对较低,与当地森林地区丰度相当;ISS中的草本、灌木、乔木已被开发为当地牲畜的饲料来源,可减少外部投入的需求,以达到保护景观和改善土壤质量的目的,对ISS进行3-15年的年序研究发现,年序越大的ISS的细菌群落越趋于相似。Yang等[54]使用基于amoA基因的Illumina Miseq高通量测序检测中国北部小麦-玉米轮作土壤中施肥种类和灌溉频率对AOA和AOB的影响,结果表明AOA群落主要受到灌溉频率的影响,而AOB对于施肥种类有更强地响应;另外,土壤湿度、pH对AOA有显著影响,总有机碳对AOB影响显著,而土壤总氮含量对AOA和AOB均有显著影响。受农业影响越大的土壤(特别是施肥的影响)中AOB的丰度和硝化潜力越大,越接近于自然生态系统的农业土壤中的AOB越接近于普通水平。AOA和AOB对农业措施的响应是不一样的,AOA群落受到灌溉频率的影响更大。
在废水处理系统中,Gao等[55]运用基于amoA基因的qPCR检测生活污水、工业污水和实验室污水处理共10个污水处理系统中AOA和AOB的丰度,发现AOB比AOA丰度更高、分布更广泛,说明在污水处理系统中AOB可能比AOA发挥更重要的作用。Zhang等[56]运用基于amoA基因和16S rRNA基因的GeoChip、454焦磷酸测序和qPCR分子检测技术,分析了螺旋霉素或者氧四环素废水处理系统中AOA和AOB的群落变化,结果表明在含有螺旋霉素的废水处理系统中AOA丰度显著高于AOB,而氧四环素系统中AOA丰度低于AOB,无抗生素的对照系统中没有检测到AOA;螺旋霉素系统中AOA的78.5%-99.6%属于奇古菌门(Thaumarchaeota);GeoChip结果显示AOA的amoA基因信号强度与螺旋霉素的浓度相关(P < 0.05),AOA在高浓度的螺旋霉素压力下比AOB更能占据优势。在不同的废水处理系统中,AOA和AOB的分布和丰度是不一样的,在生活污水、工业污水、实验室污水和氧四环素污水中AOB比AOA丰度更高,而在高浓度的螺旋霉素压力下AOA比AOB更能占据优势。
在海洋环境中,影响氨氧化微生物群落组成和分布的主要环境因素有温度、盐度、深度、水溶氧含量和碳氮含量等。Vetterli等[57]运用基于amoA基因的T-RFLP法分析发现,芬兰海湾富营养化沉积物中氨氧化微生物的丰度很高,其中AOA群落分布格局具有强烈的空间变化,而AOB群落分布则具有显著的时间变化特点,而在开放海域,AOA和AOB丰度很低,以芬兰海湾特有的物种为主;影响氨氧化微生物时空分布模式的主要环境因素有盐度、碳氮含量以及水溶氧含量。Bertagnolli等[58]运用基于amoA基因的Illumina Miseq高通量测序检测智利海岸海水中奇古菌的丰度,智利海岸海水是季节性低氧或缺氧的,其温度、盐度和深度是对奇古菌群落组成影响最大的因素;奇古菌门的Nitrosopumilus-A在所有样本中均有分布(42%-100% OTU),而Nitrosopumilus-B主要分布在春夏季节含氧量下降的深层海水中。在海洋环境中AOA和AOB的分布格局不同,AOA受空间分布影响,而AOB受时间分布影响,温度、盐度、深度等环境因素都是影响海洋氨氧化微生物的重要因子,而影响不同遗传谱系的AOA的环境因子也不同。
不管在土壤、废水还是海洋系统中,AOA和AOB的表现都会呈现出明显的差异,对两者产生影响的环境因子也不同。目前很多研究中都会使用不同的技术手段对这两者进行分别研究,但无疑更新的、更细致的技术能够更好地检测出这两者之间存在的更细微的差别,目前检测两者表现差异使用比较多的技术是Illumina Miseq高通量测序、GeoChip和qPCR[54, 56, 58]。
3.2 羟胺氧化还原酶基因hao和亚硝酸盐氧化还原酶基因nxrA羟胺氧化还原酶(Hao)催化NH2OH氧化为NO2-,hao基因是对应的功能标记基因[49]。对hao基因的研究目前主要集中于分离纯菌株及对功能蛋白和基因结构的研究[49, 59-61],对其在环境样本中的研究则鲜有报道。陈春兰等[49]以一个水稻长期定位试验田为平台,构建hao基因和amoA基因克隆文库,研究长期施氮肥对亚硝化菌和氨氧化菌多样性及其群落结构的影响。结果表明,施氮肥使氨氧化微生物和亚硝化菌多样性降低,群落结构趋于单一。
亚硝酸盐氧化还原酶(Nxr)催化NO2-氧化为NO3-。Nxr酶是由一个α亚基和一个β亚基组成的异源二聚体,nxrA基因编码Nxr酶催化亚基α亚基,是Nxr酶的功能标记基因[50]。Rani等[62]将nxrA基因作为功能标记基因进行扩增子焦磷酸测序,发现全球六个不同地域的海洋沉积物中的硝化刺菌属(Nitrospina)细菌数量分布出乎意料的少,且极地区域的多样性低于非极地区域。
在以上报道中,检测到的硝化作用相关功能基因主要分布在在农业系统土壤、废水系统和海洋环境中。amoA基因是硝化作用中最受关注的基因,研究amoA基因使用的检测技术非常多样,从Illumina Miseq高通量测序、GeoChip功能基因芯片、qPCR到DGGE、T-RFLP、克隆文库均有涉及。GeoChip功能基因芯片、qPCR均可定量,但qPCR通量偏低;Illumina Miseq高通量测序、GeoChip功能基因芯片通量高,但GeoChip功能基因芯片只能检测已知基因序列,Illumina Miseq高通量测序有读长限制以及取样、测序时随机因素和PCR扩增的影响[11]。
4 反硝化作用功能基因反硝化作用(Denitrification)从NO3-到N2包括四个过程(图 1):NO3-→NO2-、NO2-→NO、NO→N2O和N2O→N2,这些反应过程对应的功能标记基因分别是narG基因、nirK和nirS基因、norB基因以及nosZ基因。一直以来人们都认为反硝化作用只由细菌完成,然而近年来有研究表明真菌和古菌也能进行反硝化活动[63]。
4.1 硝酸盐还原酶基因narG催化NO3-还原化为NO2-的硝酸盐还原酶(Nar)由两个在胞质中的亚基NarGH、一个膜结合亚基NarI以及NarDJ组成,narG基因编码Nar酶的活性亚基NarG亚基,是Nar酶的功能标记基因[64-65]。
目前,以narG基因作为标记基因检测环境样本中的反硝化菌的研究相对而言并不特别多。Philippot等[66]构建narG基因克隆文库,对玉米的不同培养变种根际反硝化菌群落进行分析,发现环境因素比玉米变种类型对反硝化菌的群落的影响更大。Deiglmayr等[67]运用基于narG基因的PCR-RFLP法分析发现,NO3-浓度变化对硝酸盐还原菌群落功能稳定性的影响不显著,是因为Nar酶对草地土壤中NO3-浓度变化有很高的适应性。Bulc等[68]使用qPCR法研究了人工沟渠中narG基因的分布,发现无论沟渠中废水的含氧量和含氮量如何,高度波动的排水模式是影响整个废水系统中narG基因丰度和分布的主要原因。以上结果表明,在农业土壤和废水沟渠中NO3-浓度和含氮量都不是影响narG基因丰度和分布的主要原因。
4.2 亚硝酸还原酶基因nirK和nirS亚硝酸盐还原酶(Nir)催化NO2-还原化为NO。Nir酶复合体中,nirK基因编码依赖铜离子的亚基NirK,而nirS基因编码含有细胞色素cd-1的亚基NirS[65, 69, 70]。在水体环境中nirS基因的丰度和多样性显著高于nirK,但通过这两个基因得到的群落结构均存在季节性变化。Lee等[71]使用基于nirS和nirK基因的qPCR法研究反硝化菌在旧金山海湾河口沉积物中的分布,发现在河口区域nirS基因的丰度和多样性均显著高于nirK;两个基因存在相似的空间变化规律,而时间变化规律有差异;当硝酸盐浓度高时nirS丰度高,而温度低时nirK丰度高。Zhou等[72]使用基于nirS和nirK基因的Illumina Miseq高通量测序研究中国北部一个浅层富营养化的水库中反硝化菌群落结构的变化,NirS型反硝化菌的丰度和多样性高于NirK型反硝化菌;沉积物总氮和温度是影响反硝化菌群落结构季节性变化的重要环境因子。
4.3 一氧化氮还原酶基因norB一氧化氮还原酶(Nor)将NO还原为N2O,norB基因编码Nor酶复合体NorBCDEFQZ中的NorB亚基,是Nor酶的功能标记基因[65]。Fagerstone等[73]通过norB基因的PCR检测确认反硝化菌的存在,且发现抗生素处理反硝化细菌可以大大减少微藻养殖中N2O的排放量。Kearns等[74]构建norB基因和nosZ基因的克隆文库,同时使用qPCR法分析发现施氮肥的盐沼中含有norB基因和含有nosZ基因的微生物均属于α-和β-变形菌;当氮肥浓度非常高时,nosZ基因丰度显著降低,norB基因丰度没有显著变化。抗生素可以降低norB基因丰度,但高氮肥浓度对norB基因没有显著影响。
4.4 氧化亚氮还原酶基因nosZ氧化亚氮还原酶(Nos)将N2O还原为N2。Nos酶在系统发育上有两个不同的NosZ分枝,一个分枝含有典型的Z型NosZ蛋白,另一个含有非典型的NosZ蛋白[75]。编码典型的Z型NosZ蛋白的nosZ基因存在于能够完成反硝化作用的细菌中,是Nos酶的功能标记基因;非典型的nosZ基因存在于能够完成更多样化的氮代谢途径的细菌中,包括那些缺乏nirS和nirK基因的细菌[75-77]。Orellana等[75]结合宏基因组鸟枪测序法和基于nosZ基因的Illumina测序,分析了美国中西部玉米主要产区代表性的砂质和泥质土壤中nosZ基因的丰度和多样性,发现非典型的nosZ基因数量超过典型的nosZ基因,突出了非典型nosZ基因微生物在土壤中和在其他环境中消耗N2O的潜在作用。
水体环境中溶解氧浓度是影响反硝化菌的重要因素。Wyman等[78]利用qPCR方法分析了阿拉伯海富氧和弱氧的海水中进行反硝化作用的α-变形菌的nosZ基因的表达情况,发现宽阔富氧的海洋表面比弱氧的海水中的反硝化菌丰度高,富氧的海洋表面可能是N2O的潜在汇。
反硝化作用可以将环境的硝态氮肥转化为N2O和N2释放到大气中,与农业系统息息相关。在以上研究中,对反硝化作用相关功能基因使用最多的分子检测技术是qPCR。qPCR有很好的定量性,可以定量不同环境中反硝化作用功能基因的分布,但其通量低、非特异性扩增和扩增偏差也是很明显的局限性(表 1)。
5 厌氧氨氧化作用功能基因厌氧氨氧化作用(Anammox)将NH4+氧化为N2(图 1)。厌氧氨氧化作用的分子机制虽然一直未知,但Kartal等[79]提出,厌氧氨氧化作用中N2H4是由厌氧底物NH4+和NO2-产生,NO是N2H4的直接前体。hzsA基因和hzo基因均可以提供环境中厌氧氨氧化细菌的物种分类信息,甚至可能比16S rRNA基因更胜一筹,经常作为厌氧氨氧化细菌的标记基因[80-81]。
N2H4合成酶(Hzs)催化NH4+在厌氧条件下与由NO2-还原产生的NO反应生成N2H4,Hzs酶由hzsA、hzsB、hzsC基因共同编码,hzsA基因编码Hzs酶的α亚基,是Hzs酶的功能标记基因[82-83]。N2H4氧化还原酶(Hzo)催化N2H4氧化为N2,Hzo酶由hzo基因编码[84]。
厌氧氨氧化细菌分布非常广泛,在海洋、海岸和江河口的沉积物、红树林、海洋冰块和淡水湖中,甚至是西非、智利和秘鲁等氧气含量最小带(Oxygen minimum zones,OMZs)都发现有厌氧氨氧化细菌的存在[6]。Russ等[85]运用基于hzsA基因的qPCR分析加利福尼亚湾兼有冷烃富集流和热液喷口地区的沉积物,发现所有的厌氧氨氧化菌都与“Candidatus Scalindua”属相近,但系统发育上属于两个不同的hzsA基因序列簇,相似性低于76%。Bale等[86]在英格兰北海南部砂质和泥质砂岩沉积物中,通过16S rRNA和hzsA基因的逆转录qPCR和15N稳定同位素标记实验发现,厌氧氨氧化菌在有机碳含量高的沉积物中活性更高,并且在夏天比冬天活性和丰度更高。Sun等[81]构建16S rRNA基因和hzo基因克隆文库,发现NH4+与NO3-、NO2-的比例显著影响了厌氧氨氧化细菌在东江河中的垂直分布。Naeher等[84]运用基于16S rRNA基因和hzo基因的qPCR分析发现,厌氧氨氧化菌在整个法国塞纳河河口中均有分布,且丰度很高。以上研究中检测到的厌氧氨氧化菌主要分布在水体环境中,hzsA基因标记的厌氧氨氧化菌在有机碳含量高的沉积物中活性更高,而不同氮素形态的比例会显著影响hzo基因标记的厌氧氨氧化菌。
厌氧氨氧化作用功能基因一般分布在水体环境中。使用最多的分子检测技术是qPCR,可以对分布于不同水体环境中的厌氧氨氧化作用功能基因进行定量和检测(表 1)。
6 氮同化还原作用及异化还原作用功能基因氮同化还原作用(Assimilatory nitrogen reduction)及异化还原作用(Dissimilatory nitrogen reduction to ammonia,DNRA)均可将NO3-还原为NH4+,这一反应包括两个步骤:NO3-还原为NO2-、NO2-还原为NH4+(图 1)。DNRA与氮同化还原作用的不同之处在于,DNRA不需要NAD(P)H作为电子供体,且不存在有机氮中间体[87]。长久以来,DNRA在氮循环中都是一个被忽略的过程,直到Lam等[88]提出DNRA是微生物在厌氧氨氧化作用中利用的NH4+的来源。此外,最近证实一部分反硝化菌有时也可以进行DNRA,DNRA在环境中的分布可能比以往所认知地更为常见[2]。
6.1 硝酸盐同化还原酶基因nasA、narB和异化还原酶基因napA硝酸盐同化还原酶Nas和Nar催化NO3-同化还原为NO2-[64]。Nas酶大亚基由nasA基因编码,相应的功能基因是nasA基因[89],而硝酸盐同化还原酶Nar的标记基因为narB基因[90]。硝酸盐异化还原酶(Nap)催化NO3-异化还原为NO2-,napA基因编码Nap酶复合体NapAB中的NapA亚基。Feng等[91]运用PCR扩增napA基因,发现三个油井废水中napA基因的丰度和分布会随着有机碳浓度的变化而变化,才将napA基因作为异化硝酸盐还原菌的标记基因。
海洋环境中,氮同化还原菌的生物地理分布模式与环境条件密切相关。Paerl等[92]在加州湾一个由沿岸涌升流和贫营养太平洋组成的动态区域(California current system,CCS)中,运用qPCR分析发现在CCS的核心区域聚球藻的数量和聚球藻narB基因的丰度较低,而沿岸过渡地带较高;该研究组运用同样的方法分析发现,蒙特利湾每年春季上升的涌流导致聚球藻种群及其narB基因亚群丰度下降,且所检测的金属离子等环境因子对narB基因的丰度没有显著影响[93]。Jiang等[89]建立16S rRNA和nasA基因克隆文库,分析硝酸盐同化细菌在几个大洋沿岸海域的地理分布和多样性,发现盐浓度、温度和硝酸盐浓度是影响氮同化还原菌的分布和多样性的主要环境因子。
6.2 亚硝酸盐同化还原酶基因nirA、nirB和异化还原酶基因nrfA亚硝酸盐同化还原酶(Nir)催化NO2-同化还原为NH4+[94]。nirA和nirB基因分别编码NirA和NirB亚基,这两种基因可作为Nir酶的功能标记基因[95]。在多种蓝藻中已发现一种nirA操纵子,包含亚硝酸同化还原酶基因nirA、编码转运NO3-和NO2-的ABC型转运蛋白的nrtABCD基因,和硝酸盐同化还原酶基因narB,nirA操纵子只有在NO3-或NO2-浓度高而NH4+浓度低的时候才会高度表达[95]。nirB基因转录在nirA操纵子的上游,其编码的NirB酶在NirA酶成熟过程中扮演骨架蛋白的角色,NirA酶的表达需要NirB酶的存在[95-97]。Alcantara-Hernandez等[98]利用narB基因和nirA基因作为功能标记基因对墨西哥一块极端盐碱地(以前是Texcoco湖)进行扩增子测序,发现含有narB和nirA基因的嗜盐古菌占主要地位;在pH大于10甚至更高的环境中NH4+会转化成NH3,而NO3-和NO2-是更好的氮源,所以嗜盐古菌对硝酸盐的代谢能力对其生存有重要意义。
甲酸依赖型亚硝酸盐还原酶(Nrf)催化NO2-异化还原为NH4+。Nrf酶由一个7个基因组成的操纵子nrfABCDEFG编码,nrfA编码的催化亚基NrfA是利用NO2-作为电子受体的细胞色素C,nrfBCD编码的蛋白将电子转移到催化亚基NrfA上,nrfEFG编码的血红裂解酶(Heme lyase)将血红素(Heme group)连结到NrfA催化位点上[99]。nrfA基因是Nrf酶的标记基因。Song等[100]以nrfA基因为标记基因对美国北卡罗来纳州一个浅泻湖河口沉积物群落进行焦磷酸测序,发现其中DNRA活性和nrfA基因的丰度随有机质含量的增加而上升,有机碳的可获得性是DNRA群落活性的重要调节因子。
随着对DNRA的认识逐渐加深,DNRA也还渐成为研究的热点之一[2]。在氮同化还原和异化还原作用研究中,使用的分子检测技术有焦磷酸测序、qPCR、克隆文库等,目前对这两个过程的功能基因的研究还不是很多,但是随着关注度的加深也会随之变多。
7 氨化作用及同化作用氨化作用(Ammonification),也称作氮矿化作用(Nitrogen mineralization),是有机质氮转化为NH4+的过程(图 1)[101]。不同的有机质需要不同的酶催化氨化作用,其中尿素酶(Urease)催化尿素的氨化作用。尿素酶可催化尿素分解为NH4+,系统命名为酰胺水解酶[102-103],细菌、植物和动物体内都含有尿素酶,通过将尿素分解为NH4+的过程获取能量[104]。细菌尿素酶基因簇由结构基因、辅助基因和调节基因组成,结构基因包括ureA、ureB和ureC基因,分别编码α、β和γ亚基;辅助基因包括ureD、ureE、ureF、ureG、ureH、ureI等,ureR是调节基因。ureC基因编码尿素酶维持酶活性的蛋白,是尿素酶的标记基因[103]。
同化作用(Assimilation),又称为合成代谢,是指生物体利用能量将小分子合成为大分子的一系列代谢途径。所有的氮循环微生物可通过同化作用利用环境中的小分子氮化物合成含氮大分子[105]。
8 结论与展望生态系统是一个整体,而氮循环并非独立存在于生态系统中,氮元素不同形式转化过程中,会偶联其他元素的转化过程,如碳循环、硫循环等,其中厌氧甲烷氧化和反硝化作用的偶联就是非常典型的例子[106]。未来对氮循环的研究必然会发展为氮循环偶联其他生物地球化学循环的研究。但不管研究的方向如何改变,分子检测技术都是其中重要的研究手段,正如高通量测序发现完全硝化菌的过程[8-9]。单细胞测序无疑是目前最新的技术,而高通量qPCR是在普通qPCR基础上研发的高通量技术[107],虽然目前还没有这些新技术应用在氮循环研究中的实例,但在不久的将来,这些新技术必然会给氮循环研究带来新的发现。
此外,大数据时代带来的另一方面的需求就是数据分析平台的建立。随着微生物组学技术的普及,在未来数十年之内数据分析的基础平台建设将对氮循环微生物功能基因的研究提供保障,而分析技术本身的研究和发展也非常重要,基础性分析和存储平台的建设可为氮循环功能类群研究提供坚实的基础[108]。以功能基因为起点,以日益发展的数据分析平台为支撑,分子检测技术的革新能够更深入全面地了解参与氮循环的微生物功能类群多样性,最终为环境科学、生态学和地球科学的发展带来新的契机。
| [1] |
Canfield DE, Glazer AN, Falkowski PG. The evolution and future of earth's nitrogen cycle[J]. Science, 2010, 330: 192-196. DOI:10.1126/science.1186120 |
| [2] |
Nelson MB, Martiny AC, Martiny JB. Global biogeography of microbial nitrogen-cycling traits in soil[J]. Proc Natl Acad Sci USA, 2016, 113: 8033-8040. DOI:10.1073/pnas.1601070113 |
| [3] |
Jetten MS. The microbial nitrogen cycle[J]. Environ Microbiol, 2008, 10(11): 2903-2909. DOI:10.1111/emi.2008.10.issue-11 |
| [4] |
Venter JC RK, Heidelberg JF, Halpern AL, et al. Environmental genome shotgun sequencing of the sargasso sea[J]. Science, 2004, 304: 66-74. DOI:10.1126/science.1093857 |
| [5] |
Konneke M, Bernhard AE, de la Torre JR, et al. Isolation of an autotrophic ammonia-oxidizing marine archaeon[J]. Nature, 2005, 437: 543-546. DOI:10.1038/nature03911 |
| [6] |
He JZ, Zhang LM. Advances in ammonia-oxidizing microorganisms and global nitrogen cycle[J]. Acta Ecologica Sinica, 2009, 29: 406-415. |
| [7] |
Monteiro M, Seneca J, Magalhaes C. The history of aerobic ammonia oxidizers:from the first discoveries to today[J]. J Microbiol, 2014, 52: 537-47. DOI:10.1007/s12275-014-4114-0 |
| [8] |
Daims H, Lebedeva EV, Pjevac P, et al. Complete nitrification by nitrospira bacteria[J]. Nature, 2015, 528: 504-509. DOI:10.1038/nature16461 |
| [9] |
van Kessel MA, Speth DR, Albertsen M, et al. Complete nitrification by a single microorganism[J]. Nature, 2015, 528: 555-559. DOI:10.1038/nature16459 |
| [10] |
Baptista JD, Lunn M, Davenport RJ, et al. Agreement between amoA gene-specific quantitative PCR and fluorescence in situ hybridization in the measurement of ammonia-oxidizing bacteria in activated sludge[J]. Appl Environ Microbiol, 2014, 80: 5901-5910. DOI:10.1128/AEM.01383-14 |
| [11] |
Deng Y, Feng K, Wei ZY, et al. Recent studies and applications of metagenomics in environmental engineering[J]. Chinese Journal of Environmental Engineering, 2016, 10: 3373-3382. |
| [12] |
Liu YY, Wang S, Li SZ, et al. Advances in molecular ecology on microbial functional genes of carbon cycle[J]. Microbiology China, 2017, 44: 1676-1689. |
| [13] |
Bustin S, Benes V, Garson J, et al. The miqe guidelines:minimum information for publication of quantitative real-time pcr experiments[J]. Clinical Chemistry, 2009, 55: 611-622. DOI:10.1373/clinchem.2008.112797 |
| [14] |
Jia ZJ. Principle and application of DNA-based stable isotope Probing—A review[J]. Acta Microbiologica Sinica, 2011, 51: 1585-1594. |
| [15] |
Ge Y, He JZ, Zheng YM, et al. Stable isotope probing and its applications in microbial ecology[J]. Acta Ecologica Sinica, 2006, 26: 1574-1582. |
| [16] |
He SB, Chai LQ, Tan JJ, et al. Rencent advance in fluorescence in situ hybridization[J]. Plant Science Journal, 2014, 32: 199-204. |
| [17] |
Lin H, Fang SG. Genomic library construction and perspectives on applications in conservation genetics[J]. Acta Theriologica Sinica, 2005, 25: 86-90. |
| [18] |
Zhu ZX, Chen X. Single cell sequencing technology and its applications progress[J]. Genomics and Applied Biology, 2015, 34: 000902-908. |
| [19] |
Blainey PC. The future is now:single-cell genomics of bacteria and archaea[J]. FEMS Microbiol Rev, 2013, 37: 407-427. DOI:10.1111/1574-6976.12015 |
| [20] |
Tu Q, Yu H, He Z, et al. Geochip 4:A functional gene-array-based high-throughput environmental technology for microbial community analysis[J]. Mol Ecol Resour, 2014, 14: 914-928. |
| [21] |
Spencer SJ, Tamminen MV, Preheim SP, et al. Massively parallel sequencing of single cells by epicpcr links functional genes with phylogenetic markers[J]. ISME J, 2016, 10: 427-436. DOI:10.1038/ismej.2015.124 |
| [22] |
Schmidt TM, Delong EF, Pace NR. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing[J]. Journal of Bacteriology, 1991, 173: 4371. DOI:10.1128/jb.173.14.4371-4378.1991 |
| [23] |
Manz W, Szewzyk U, Ericsson P, et al. In situ identification of bacteria in drinking water and adjoining biofilms by hybridization with 16S and 23S rRNA-Directed fluorescent oligonucleotide probes[J]. Applied & Environmental Microbiology, 1993, 59: 2293-2298. |
| [24] |
Muyzer G, de Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA[J]. Appl Environ Microbiol, 1993, 59: 695-700. |
| [25] |
Liu WT, Marsh TL, Cheng H, et al. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA[J]. Applied & Environmental Microbiology, 1997, 63: 4516-4522. |
| [26] |
Radajewski S, Ineson P, Parekh NR, et al. Stable-isotope probing as a tool in microbial ecology[J]. Nature, 2000, 403: 646-649. DOI:10.1038/35001054 |
| [27] |
Chandler DP, Stults JR, Cebula S, et al. Affinity purification of DNA and RNA from environmental samples with peptide nucleic acid clamps[J]. Applied & Environmental Microbiology, 2000, 66: 3438-3445. |
| [28] |
Wu L, Thompson DK, et al. Development and evaluation of functional gene arrays for detection of selected genes in the environment[J]. Appl Environ Microbiol, 2001, 67: 5780-5790. DOI:10.1128/AEM.67.12.5780-5790.2001 |
| [29] |
Poinar HN, Schwarz C, Qi J, et al. Metagenomics to paleogenomics:Large-scale sequencing of mammoth DNA[J]. Science, 2006, 311: 392-394. DOI:10.1126/science.1123360 |
| [30] |
Edwards RA, Rodriguez-Brito B, Wegley L, et al. Using pyrosequencing to shed light on deep mine microbial ecology[J]. BMC Genomics, 2006, 7: 57. DOI:10.1186/1471-2164-7-57 |
| [31] |
Yoon HS, Price DC, Stepanauskas R, et al. Single-cell genomics reveals organismal interactions in uncultivated marine protists[J]. Science, 2011, 332: 714-717. DOI:10.1126/science.1203163 |
| [32] |
Novak R, Zeng Y, Shuga J, et al. Single-cell multiplex gene detection and sequencing with microfluidically generated agarose emulsions[J]. Angew Chem Int Ed Engl, 2011, 50: 390-395. DOI:10.1002/anie.201006089 |
| [33] |
Rotthauwe JH, Witzel KP, Liesack W. The ammonia monooxygenase structural gene amoA as a functional marker:molecular fine-scale analysis of natural ammonia-oxidizing populations[J]. Applied & Environmental Microbiology, 1997, 63: 4704. |
| [34] |
Purkhold U, Pommereningröser A, Juretschko S, et al. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis:implications for molecular diversity surveys[J]. Applied & Environmental Microbiology, 2000, 66: 5368-5382. |
| [35] |
Horz HP, Rotthauwe JH, Lukow T, et al. Identification of major subgroups of ammonia-oxidizing bacteria in environmental samples by T-RFLP analysis of amoA PCR products[J]. Journal of Microbiological Methods, 2000, 39: 197-204. DOI:10.1016/S0167-7012(99)00119-0 |
| [36] |
Oved T, Shaviv A, Goldrath T, et al. Influence of effluent irrigation on community composition and function of ammonia-oxidizing bacteria in soil[J]. Appl Environ Microbiol, 2001, 67: 3426-3433. DOI:10.1128/AEM.67.8.3426-3433.2001 |
| [37] |
Harms G, Layton AC, Dionisi HM, et al. Real-Time PCR quantification of nitrifying bacteria in a municipal wastewater treatment plant[J]. Environmental Science & Technology, 2003, 37: 343-351. |
| [38] |
Freitag TE, Chang L, Prosser JI. Changes in the community structure and activity of betaproteobacterial ammonia-oxidizing sediment bacteria along a freshwater-marine gradient[J]. Environ Microbiol, 2006, 8: 684-696. DOI:10.1111/emi.2006.8.issue-4 |
| [39] |
Yergeau E, Hogues H, Whyte LG, et al. The functional potential of high arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses[J]. ISME J, 2010, 4: 1206-1214. DOI:10.1038/ismej.2010.41 |
| [40] |
Gaby JC, Buckley DH. A Comprehensive aligned nifh gene database:a multipurpose tool for studies of nitrogen-fixing bacteria[J]. Database(Oxford), 2014, 2014: bau001. |
| [41] |
Raymond J, Siefert JL, Staples CR, et al. The natural history of nitrogen fixation[J]. Mol Biol Evol, 2004, 21: 541-554. DOI:10.1093/molbev/msh047 |
| [42] |
Soni R, Suyal DC, Sai S, et al. Exploration of nifh gene through soil metagenomes of the western indian himalayas[J]. Biotech, 2016, 6: 1-4. |
| [43] |
Chien Y T, Zinder SH. Cloning, DNA sequencing, and characterization of a nifD-homologous gene from the archaeon Methanosarcina barkeri 227 which resembles nifD1 from the eubacterium Clostridium pasteurianum[J]. Journal of Bacteriology, 1994, 176: 6590-6598. DOI:10.1128/jb.176.21.6590-6598.1994 |
| [44] |
Wang J, Bao JT, Li XR, et al. Molecular ecology of nifh genes and transcripts along a chronosequence in revegetated areas of the tengger desert[J]. Microb Ecol, 2016, 71: 150-163. DOI:10.1007/s00248-015-0657-9 |
| [45] |
Wang J, Zhang D, Zhang L, et al. Temporal variation of diazotrophic community abundance and structure in surface and subsoil under four fertilization regimes during a wheat growing season[J]. Agriculture, Ecosystems & Environment, 2016, 216: 116-124. |
| [46] |
Zhang B, Penton CR, Xue C, et al. Evaluation of the ion torrent personal genome machine for gene-targeted studies using amplicons of the nitrogenase gene nifh[J]. Appl Environ Microbiol, 2015, 81: 4536-4545. DOI:10.1128/AEM.00111-15 |
| [47] |
Wang L, Yu Z, Yang J, et al. Diazotrophic bacterial community variability in a subtropical deep reservoir is correlated with seasonal changes in nitrogen[J]. Environ Sci Pollut Res Int, 2015, 22: 19695-19705. DOI:10.1007/s11356-015-5144-9 |
| [48] |
Xiao P, Jiang Y, Liu Y, et al. Re-evaluation of the diversity and distribution of diazotrophs in the south china sea by pyrosequencing the nifh gene[J]. Marine and Freshwater Research, 2015, 66: 681. DOI:10.1071/MF14134 |
| [49] |
Chen CL, Wu MN, Wei WX. Effect of long-term application of nitrogen fertilizer on the diversity of nitrifying genes(amoa and hao)in paddy soil[J]. Environmental Science, 2011, 32: 1489-1496. |
| [50] |
Wertz S, Poly F, Le Roux X, et al. Development and application of a PCR-denaturing gradient gel electrophoresis tool to study the diversity of Nitrobacter-like nxrA sequences in soil[J]. FEMS Microbiol Ecol, 2008, 63: 261-271. DOI:10.1111/fem.2008.63.issue-2 |
| [51] |
Avrahami S, Conrad R. Cold-temperate climate:a factor for selection of ammonia oxidizers in upland soil?[J]. Can J Microbiol, 2005, 51: 709-714. DOI:10.1139/w05-045 |
| [52] |
Purkhold U, Pommerening-Röser A, Juretschko S, et al. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis:implications for molecular diversity surveys[J]. Applied & Environmental Microbiology, 2001, 5368-5382. |
| [53] |
Cubillos AM, Vallejo VE, Arbeli Z, et al. Effect of the conversion of conventional pasture to intensive silvopastoral systems on edaphic bacterial and ammonia oxidizer communities in Colombia[J]. European Journal of Soil Biology, 2016, 72: 42-50. DOI:10.1016/j.ejsobi.2015.12.003 |
| [54] |
Yang YD, Ren YF, Wang XQ, et al. Ammonia-oxidizing archaea and bacteria responding differently to fertilizer type and irrigation frequency as revealed by Illumina Miseq sequencing[J]. Journal of Soils and Sediments, 2017, 1-12. |
| [55] |
Gao J, Luo X, Wu G, et al. Abundance and diversity based on amoA genes of ammonia-oxidizing archaea and bacteria in ten astewater treatment systems[J]. Applied Microbiology & Biotechnology, 2013, 98: 3339-3354. |
| [56] |
Zhang Y, Tian Z, Liu M, et al. High Concentrations of the antibiotic spiramycin in wastewater lead to high abundance of ammonia-oxidizing archaea in nitrifying populations[J]. Environ Sci Technol, 2015, 49: 9124-132. DOI:10.1021/acs.est.5b01293 |
| [57] |
Vetterli A, Hietanen S, Leskinen E. Spatial and temporal dynamics of ammonia oxidizers in the sediments of the gulf of Finland, Baltic Sea[J]. Mar Environ Res, 2016, 113: 153-63. DOI:10.1016/j.marenvres.2015.12.008 |
| [58] |
Bertagnolli AD, Ulloa O. Hydrography shapes amoA containing thaumarcheota in the coastal waters off central chile[J]. Environ Microbiol Rep, 2017, 9(6): 717-728. DOI:10.1111/emi4.2017.9.issue-6 |
| [59] |
Hiroya S MF. Comparison of 16S rRNA, ammonia monooxygenase subunit A and hydroxylamine oxidoreductase gene, in chemolithotrophic ammonia-oxidizing bacteria[J]. Journal of General & Applied Microbiology, 2002, 48: 173-176. |
| [60] |
Moran MA, Buchan A, González JM, et al. Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment[J]. Nature, 2004, 432: 910-913. DOI:10.1038/nature03170 |
| [61] |
Poret-Peterson AT, Graham JE, Gulledge J, et al. Transcription of nitrification genes by the methane-oxidizing bacterium, Methylococcus capsulatus strain Bath[J]. ISME J, 2008, 2: 1213-1220. DOI:10.1038/ismej.2008.71 |
| [62] |
Rani S, Koh HW, Rhee SK, et al. Detection and diversity of the nitrite oxidoreductase alpha subunit(nxrA)gene of nitrospina in marine sediments[J]. Microb Ecol, 2017, 73: 111-122. DOI:10.1007/s00248-016-0897-3 |
| [63] |
Shoun H, Fushinobu S, Jiang L, et al. Fungal denitrification and nitric oxide reductase cytochrome P450nor[J]. Philos Trans R Soc Lond B Biol Sci, 2012, 367: 1186-1194. DOI:10.1098/rstb.2011.0335 |
| [64] |
Reyna L, Wunderlin DA, Genti-Raimondi S. Identification and quantification of a novel nitrate-reducing community in sediments of suquia river basin along a nitrate gradient[J]. Environ Pollut, 2010, 158: 1608-1614. DOI:10.1016/j.envpol.2009.12.014 |
| [65] |
Zumft WG. Cell biology and molecular basis of denitrification[J]. Microbiology & Molecular Biology Reviews, 1997, 61: 533-616. |
| [66] |
Philippot L, Kuffner M, et al. Genetic structure and activity of the nitrate-reducers community in the rhizosphere of different cultivars of maize[J]. Plant and Soil, 2006, 287: 177-186. DOI:10.1007/s11104-006-9063-x |
| [67] |
Deiglmayr K, Philippot L, Kandeler E. Functional stability of the nitrate-reducing community in grassland soils towards high nitrate supply[J]. Soil Biology and Biochemistry, 2006, 38: 2980-2984. DOI:10.1016/j.soilbio.2006.04.034 |
| [68] |
Bulc TG, Klemenčič AK, Razinger J. Vegetated ditches for treatment of surface water with highly fluctuating water regime[J]. Water Science & Technology, 2011, 63: 2353. |
| [69] |
Glockner AB, Jüngst A, Zumft WG. Copper-containing nitrite reductase from pseudomonas aureofaciens is functional in a mutationally cytochrome cd1-free background(Nirs-)of Pseudomonas stutzeri[J]. Archives of Microbiology, 1993, 160: 18-26. |
| [70] |
Yang JK, Cheng ZB, Li J, et al. Community composition of nirS-type denitrifier in a shallow eutrophic lake[J]. Microb Ecol, 2013, 66: 796-805. DOI:10.1007/s00248-013-0265-5 |
| [71] |
Lee JA, Francis CA. Spatiotemporal characterization of San Francisco bay denitrifying communities:A comparison of nirK and nirS diversity and abundance[J]. Microb Ecol, 2017, 73: 271-284. DOI:10.1007/s00248-016-0865-y |
| [72] |
Zhou S, Huang T, et al. Illumina Miseq sequencing reveals the community composition of Nirs-Type and Nirk-Type denitrifiers in zhoucun Reservoir -a large shallow eutrophic reservoir in northern China[J]. RSC Adv, 2016, 6: 91517-91528. DOI:10.1039/C6RA18017E |
| [73] |
Fagerstone KD, Quinn JC, Bradley TH, et al. Quantitative measurement of direct nitrous oxide emissions from microalgae cultivation[J]. Environ Sci Technol, 2011, 45: 9449-9456. DOI:10.1021/es202573f |
| [74] |
Kearns PJ, Angell JH, Feinman SG, et al. Long-term nutrient addition differentially alters community composition and diversity of genes that control nitrous oxide flux from salt marsh sediments[J]. Estuarine, Coastal and Shelf Science, 2015, 154: 39-47. DOI:10.1016/j.ecss.2014.12.014 |
| [75] |
Orellana LH, Rodriguez-R LM, Higgins S, et al. Detecting nitrous oxide reductase(Nosz)genes in soil metagenomes:method development and implications for the nitrogen cycle[J]. Mbio, 2014, 5(3): e01193-14. |
| [76] |
Sanford RA, Wagner DD, Wu Q, et al. Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils[J]. Proc Natl Acad Sci USA, 2012, 109: 19709-19714. DOI:10.1073/pnas.1211238109 |
| [77] |
Jones CM, Graf DR, Bru D, et al. The unaccounted yet abundant nitrous oxide-reducing microbial community:A potential nitrous oxide sink[J]. ISME J, 2013, 7: 417-426. DOI:10.1038/ismej.2012.125 |
| [78] |
Wyman M, Hodgson S, Bird C. Denitrifying alphaproteobacteria from the arabian sea that express nosz, the gene encoding nitrous oxide reductase, in oxic and suboxic waters[J]. Appl Environ Microbiol, 2013, 79: 2670-81. DOI:10.1128/AEM.03705-12 |
| [79] |
Kartal B, Maalcke WJ, de Almeida NM, et al. Molecular mechanism of anaerobic ammonium oxidation[J]. Nature, 2011, 479: 127-30. DOI:10.1038/nature10453 |
| [80] |
Dang H, Zhou H, Zhang Z, et al. Molecular detection of candidatus scalindua pacifica and environmental responses of sediment anammox bacterial community in the Bohai sea, China[J]. PLoS One, 2013, 8: e61330. DOI:10.1371/journal.pone.0061330 |
| [81] |
Sun W, Xia C, Xu M, et al. Diversity and distribution of planktonic anaerobic ammonium-oxidizing bacteria in the Dongjiang river, China[J]. Microbiol Res, 2014, 169: 897-906. DOI:10.1016/j.micres.2014.05.003 |
| [82] |
Harhangi HR, Le Roy M, van Alen T, et al. Hydrazine synthase, a unique phylomarker with which to study the presence and biodiversity of anammox bacteria[J]. Appl Environ Microbiol, 2012, 78: 752-758. DOI:10.1128/AEM.07113-11 |
| [83] |
Shen LD, Wu HS, Gao ZQ, et al. Evidence for anaerobic ammonium oxidation process in freshwater sediments of aquaculture ponds[J]. Environmental Science and Pollution Research, 2016, 23: 1344. DOI:10.1007/s11356-015-5356-z |
| [84] |
Naeher S, Huguet A, Roose-Amsaleg CL, et al. Molecular and geochemical constraints on anaerobic ammonium oxidation(anammox)in a riparian zone of the Seine Estuary(France)[J]. Biogeochemistry, 2015, 123: 237-250. DOI:10.1007/s10533-014-0066-z |
| [85] |
Russ L, Kartal B, et al. Presence and diversity of anammox bacteria in cold hydrocarbon-rich seeps and hydrothermal vent sediments of the Guaymas Basin[J]. Front Microbiol, 2013, 4: 219. |
| [86] |
Bale NJ, Villanueva L, Fan H, et al. Occurrence and activity of anammox bacteria in surface sediments of the southern north sea[J]. FEMS Microbiol Ecol, 2014, 89: 99-110. DOI:10.1111/fem.2014.89.issue-1 |
| [87] |
Gardner WS, McCarthy MJ, An S, et al. Nitrogen fixation and dissimilatory nitrate reduction to ammonium(DNRA)support nitrogen dynamics in Texas estuaries[J]. Limnology & Oceanography, 2006, 51: 558-568. |
| [88] |
Lam P, Lavik G, Jensen MM, et al. Revising the nitrogen cycle in the peruvian oxygen minimum zone[J]. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106: 4752-4757. DOI:10.1073/pnas.0812444106 |
| [89] |
Jiang X, Dang H, Jiao N. Ubiquity and diversity of heterotrophic bacterial nasA genes in diverse marine environments[J]. PLoS One, 2015, 10: e0117473. DOI:10.1371/journal.pone.0117473 |
| [90] |
Rubio LM, Herrero A, Flores E. A cyanobacterial narB gene encodes a ferredoxin-dependent nitrate reductase[J]. Plant Molecular Biology, 1996, 30: 845-850. DOI:10.1007/BF00019017 |
| [91] |
Feng WW, Liu JF, Gu JD, et al. Nitrate-reducing community in production water of three oil reservoirs and their responses to different carbon sources revealed by nitrate-reductase encoding gene(napA)[J]. International Biodeterioration & Biodegradation, 2011, 65: 1081-1086. |
| [92] |
Paerl RW, Johnson KS, Welsh RM, et al. Differential distributions of synechococcus subgroups across the california current system[J]. Front Microbiol, 2011, 2: 59. |
| [93] |
Paerl RW, Turk KA, Beinart RA, et al. Seasonal change in the abundance of Synechococcus and multiple distinct phylotypes in Monterey Bay determined by rbcl and narB quantitative PCR[J]. Environ Microbiol, 2012, 14: 580-593. DOI:10.1111/emi.2012.14.issue-3 |
| [94] |
Buxens M, Llama MJ, Serra JL. Effect of the inorganic nitrogen source in the expression of nitrite reductase(Nira)in Bp-1[J]. Advances in Microbiology, 2014, 4(15): 1044-1056. DOI:10.4236/aim.2014.415115 |
| [95] |
Frias JE, Flores E. Induction of the nitrate assimilation nirA operon and protein-protein interactions in the maturation of nitrate and nitrite reductases in the cyanobacterium Anabaena sp. strain PCC 7120[J]. J Bacteriol, 2015, 197: 2442-2452. DOI:10.1128/JB.00198-15 |
| [96] |
Suzuki I, Horie N, Sugiyama T, Omata T. Identification and characterization of two nitrogen-regulated genes of the cyanobacterium Synechococcus sp. strain PCC7942 required for maximum efficiency of nitrogen assimilation[J]. Journal of Bacteriology, 1995, 177: 290-296. DOI:10.1128/jb.177.2.290-296.1995 |
| [97] |
Frias JE, Flores E. Negative regulation of expression of the nitrate assimilation nirA operon in the heterocyst-forming cyanobacterium Anabaena sp. strain PCC 7120[J]. J Bacteriol, 2010, 192: 2769-2778. DOI:10.1128/JB.01668-09 |
| [98] |
Alcantara-Hernandez RJ, Valenzuela-Encinas C, Zavala-Diaz de la Serna FJ, et al. Haloarchaeal assimilatory nitrate-reducing communities from a saline alkaline soil[J]. FEMS Microbiol Lett, 2009, 298: 56-66. DOI:10.1111/fml.2009.298.issue-1 |
| [99] |
Boucher DJ, Adler B, Boyce JD. The pasteurella multocida nrfE gene is upregulated during infection and is essential for nitrite reduction but not for virulence[J]. J Bacteriol, 2005, 187: 2278-2285. DOI:10.1128/JB.187.7.2278-2285.2005 |
| [100] |
Song B, Lisa JA, Tobias CR. Linking DNRA community structure and activity in a shallow lagoonal estuarine system[J]. Front Microbiol, 2014, 5: 460. |
| [101] |
Zhang X, Liu W, Schloter M, et al. Response of the abundance of key soil microbial nitrogen-cycling genes to multi-factorial global changes[J]. PLoS One, 2013, 8: e76500. DOI:10.1371/journal.pone.0076500 |
| [102] |
Mobley HL, Island MD, et al. Molecular biology of microbial ure-ases[J]. Microbiological Reviews, 1995, 59(3): 451-480. |
| [103] |
Zhao SG, Wang JQ, Bu DP, et al. Biochemistry and molecular biology of bacterial ureases[J]. Microbiology China, 2008, 35: 1146-1152. |
| [104] |
Berges JA, Contents MM. Enzymes and N cycling[J]. Nitrogen in the Marine Environment, 2008. |
| [105] |
Bolster DMWG. Glossary of terms used in bioinorganic chemistry(Iupac Recommendations 1997)[J]. Pure & Applied Chemistry, 1997, 69: 1251-1304. |
| [106] |
Raghoebarsing AA, Pol A, van de Pas-Schoonen KT, et al. A microbial consortium couples anaerobic methane oxidation to denitrification[J]. Nature, 2006, 440: 918-921. DOI:10.1038/nature04617 |
| [107] |
Schmittgen TD. High-throughput real-time PCR[J]. Methods in Molecular Biology, 2008, 429: 89. DOI:10.1007/978-1-60327-040-3 |
| [108] |
Wei ZY, Jin DC, Deng Y. Bioinformatics tools and applications in the study of environmental microbial metagenomics[J]. Microbiology China, 2015, 42(5): 890-901. |