




菌丝霉素是第一例被报道的真菌防御素,其抗菌机制清晰,药物代谢动力学研究透彻,对人的细胞无毒害作用,且能通过微生物重组制备,还可借助已经解析的三维结构对其抗菌活性进行改造提升[1-3]。鉴于此,菌丝霉素被公推为最具有临床应用潜力的三大抗菌肽之一[4]。
菌丝霉素与细菌细胞壁前体Lipid Ⅱ以1:1的化学计量结合,阻止病菌细胞壁的形成,从而阻止病菌繁殖。独特的作用机制促使了菌丝霉素不易诱导产生耐药性菌株[5]。NMR三维结构分析显示,G6、W8、D9、A31、K32、G33、G34、F35、V36和C37这10个残基参与了菌丝霉素与Lipid Ⅱ中的焦磷酸盐形成氢键[5]。菌丝霉素的作用机制明晰,为其进一步功能性改造优化奠定了理论基础[5]。Schneider等[5]通过单点饱和突变实验,证实了D12、Y29和G33对菌丝霉素的功能至关重要,不能更改替换;A31和K38仅能做同类氨基酸替换,即:A31-G;K38-R。有趣的是,张琪等[6]通过易错PCR定向进化菌丝霉素,获得了抑菌活性增强的突变体A31R。Cao等[7]将菌丝霉素氨基酸序列进行替换改造(D9-S、D11-K、Q14-H和V36-I),将正电荷氨基酸和疏水氨基酸比例提高,获得了抗菌活力增强的菌丝霉素衍生物MP1106。除此之外,菌丝霉素改造后的类似物NZ2114、AgPlectasin(rAgP)和MP1102对金黄色葡萄球菌(Staphylococcus aureus)具有较高的专一性,抗菌效果等同于甚至优于现有的青霉素和万古霉素[8-10]。
从原始真菌中提取或化学合成菌丝霉素均面临着工艺复杂、产率低或成本高等问题。已有多个学者对构建基因工程菌生产菌丝霉素进行了研究,包括原核表达和真核表达。Wang[9, 11-13]实验室利用毕赤酵母表达系统重组制备了菌丝霉素及其衍生物,产量达到了克级每升,为其产业化奠定了坚实的基础,但其稳定性和半衰期较差,无法满足实际应用需求。与此同时,Zhang[14]团队使用大肠杆菌表达系统,将菌丝霉素与Trx-tag融合表达,经Factor Xa酶切,酶切效率较低,摇瓶发酵得到3.5 mg/L纯菌丝霉素,产率仅为理论得率的20%。Chen等[15]系统研究了菌丝霉素与蛋白标签TrxA、SUMO、Intein和GST融合表达,通过蛋白酶酶切将菌丝霉素与标签蛋白分离,酶切不完全,菌丝霉素产量较低。Chen[16]实验室采用大肠杆菌系统重组表达菌丝霉素,使用凝血酶酶切除去GST标签蛋白,酶切效率较低,目标产物摇瓶发酵产量为1.6 mg/L。大肠杆菌作为实验室常用的表达系统,具有培养周期短,操作简便等优点,为了便于对菌丝霉素进行功能性改造升级,提高目标产物与标签蛋白的分离效率,急需建立一个针对菌丝霉素的高效的大肠杆菌重组表达系统。本研究首次拟将菌丝霉素衍生物MP1106与人工设计的生物表面活性剂DAMP4基因融合,使用大肠杆菌诱导表达,经包涵体复性纯化,使用本实验室制备的高活性烟草蚀纹病毒蛋白酶(Tobacco Etch Virus,TEV)酶切分割,纯化制备出高纯度抗菌肽MP1106,旨为菌丝霉素后续改造进化实验建立高效、快捷的大肠杆菌表达系统平台。
1 材料与方法 1.1 材料 1.1.1 菌种与质粒菌种E.coli BL21(DE3) 和质粒pET-28a均保存于中国科学院天津工业生物技术研究所蛋白质工程实验室。
1.1.2 试剂卡那霉素、异丙基硫代-β-D-半乳糖苷(IPTG)、β-巯基乙醇、二硫苏糖醇(DTT)购自北京索莱宝科技有限公司;烟草蚀纹病毒蛋白酶由本实验室重组制备;BCA蛋白定量试剂盒购买于北京康为世纪生物科技有限公司;目的基因MP1106通过中国科学院天津工业生物技术研究所技术支撑平台全基因合成;Ni2+-NTA亲和层析填料购买于美国GE公司;本实验所使用的其它化学试剂均为分析纯。
1.2 方法 1.2.1 DAMP4-MP1106融合蛋白的基因设计与合成据文献报道[17-19],将具有耐高温(90℃,30 min)、溶解度高、可调控等特点的人工设计的生物表面活性剂DAMP4开发成重组蛋白纯化制备的融合标签,与MP1106氨基末端融合,并在DAMP4与MP1106中间插入TEV识别位点序列,蛋白纯化后经TEV酶切,便可获得抗菌肽MP1106。为了便于目的蛋白的Western-blot检测和Ni2+-NTA纯化,在DAMP4氨基末端融合了6个His残基。最后将融合蛋白基因送至中科院天津工业生物技术研究所技术支撑平台按照大肠杆菌表达系统进行密码子优化和全基因合成,并采用EcoR I/Xho I将融合蛋白基因插入pET-28a表达载体。目的蛋白基因设计图示及序列如图 1所示。

|
| 图 1 DAMP4-MP1106融合蛋白基因的设计及序列 |
将合成的质粒pET-28a(His6-DAMP4-TEV-MP1106) 转化进E.coli BL21(DE3),使用TB培养基37℃培养OD至0.6。TB培养基配方如下:1.2%(W/V)tryptone;2.4%(W/V)yeast extract;0.4%(V/V)glycero;17 mmol/L KH2PO4;72 mmol/L KH2PO4。加入IPTG,终浓度为1 mmol/L,16℃过夜诱导表达。使用落地离心机4℃,5 000×g离心20 min,收集菌体。按照1:10(W/V)使用高盐缓冲液(50 mmol/L磷酸氢二钠-柠檬酸缓冲液,500 mmol/L Na2SO4,pH6.0) 悬浮,90℃加热30 min。4℃,12 000×g离心40 min,使用SDS-PAGE(NuPAGEs Novex 4-12%Bis-Trisgels)电泳检测上清和沉淀。
1.2.3 抗菌肽MP1106纯化制备工艺鉴于DAMP4-MP1106融合蛋白以包涵体的形式在宿主体内大量表达和积累,建立了抗菌肽MP1106的包涵体复性、纯化制备工艺流程图,如图 2所示。

|
| 图 2 抗菌肽MP1106的纯化制备工艺 |
将离心收集到的菌体使用裂解缓冲液(50 mmol/L磷酸氢二钠-柠檬酸缓冲液;150 mmol/L NaCl;pH6.0) 按照1:10(W:V)比例悬浮;使用高压匀浆破碎仪,700 bar破碎3个循环;使用落地离心机4℃,12 000×g离心40 min分离上清和沉淀;使用等体积的2% Trition-X-100冰水溶液悬浮、洗涤沉淀;在裂解缓冲液中加入尿素,配置终浓度依次为3-8 mol/L的变性缓冲液,按照1:5(W:V)比例分别悬浮、溶解沉淀,4℃搅拌30 min;12 000×g离心40 min分离上清和沉淀。使用SDS-PAGE对上述实验流程中得到的样品进行电泳检测分析。
使用优化出来的变性剂尿素浓度溶解包涵体,高速离心,分离收集上清;与使用同样缓冲液平衡后的Ni2+-NTA填料混合,4℃孵育1 h;转入重力柱分离未结合的杂蛋白,加入5倍柱体积含有30 mmol/L咪唑的洗涤缓冲液进行洗涤;配制含有300 mmol/L咪唑的洗脱缓冲液,一次一个柱体积,洗涤4次。将上述每步收集到的样品进行SDS-PAGE电泳检测分析,将含有DAMP4-MP1106融合蛋白且纯度较高的洗脱液进行合并混合。
配置pH1-12的系列缓冲液(氯化钾-盐酸缓冲液:pH1、pH2;磷酸氢二钠-柠檬酸缓冲液:pH3、pH4、pH5、pH6、pH7;Tris-盐酸缓冲液:pH8;甘氨酸-氢氧化钠缓冲液:pH9;碳酸钠-氢氧化钠缓冲液:pH10;磷酸氢二钠-氢氧化钠缓冲液:pH 11;氯化钾-氢氧化钠缓冲液:pH12)。使用96孔板按照180:20的比例使用系列pH的缓冲液10倍稀释DAMP4-MP1106融合蛋白样品,使用酶标仪测定每个样品孔中的A310值。选取浑浊度最低的缓冲液作为复性缓冲液,将DAMP4-MP1106融合蛋白进行4℃过夜透析复性。
1.2.5 TEV酶切His6-DAMP4-TEV-MP1106融合蛋白将45 mL DAMP4-MP1106融合蛋白水溶液与5 mL的10×TEV酶切缓冲液(500 mmol/L Tris-HCl;50 mmol/L EDTA;26 mmol/L β巯基乙醇;10 mmol/L DTT;pH8.0) 混合,加入0.5 mL TEV蛋白酶,室温酶切2 h。转至4℃层析柜,在裂解缓冲液中透析过夜。按照上述Ni2+-NTA亲和层析方法进行二次纯化,去除His6-DAMP4标签和TEV酶(氨基末端融合有His6标签),得到高纯度抗菌肽MP1106。
1.2.6 抗菌肽MP1106分子内二硫键的鉴定本研究通过在SDS-PAGE电泳制样缓冲液中加入20 mmol/L DTT,破坏DAMP4 -MP1106融合蛋白和抗菌肽MP1106分子内的二硫键,使其分子结构松散,SDS-PAGE电泳迁移率降低,表观分子量变大,以此初步鉴定MP1106分子内的二硫键是否正确形成。
2 结果 2.1 DAMP4-MP1106融合蛋白的诱导表达及加热处理使用SDS-PAGE电泳检测分析MP1106融合蛋白诱导表达情况,结果如图 3泳道3所示,25 kD和15 kD Mark条带间有条明显的诱导条带,分子量和DAMP4-MP1106融合蛋白分子量(20.0 kD)相符。DAMP4-MP1106融合蛋白经高盐高温处理后,离心分离得到的上清和沉淀,SDS-PAGE电泳结果如图 3泳道4和泳道5所示,全部蛋白均存在于沉淀样品中。

|
| 图 3 SDS-PAGE电泳检测DAMP4-MP1106融合蛋白的诱导表达和加热处理 M:protein ladder;1:空BL21(DE3);2:诱导前全菌;3:诱导后全菌;4:加热处理后离心上清;5:加热处理后离心沉淀 |
将诱导后菌体进行高压匀浆破碎,使用SDS-PAGE电泳检测分析DAMP4-MP1106融合蛋白可溶性表达情况。结果如图 4泳道4和泳道5,DAMP4-MP1106融合蛋白全都以包涵体形式存在,经Bio-Rad凝胶成像仪Quantify One软件检测分析,DAMP4-MP1106融合蛋白约占总蛋白23.3%。

|
| 图 4 DAMP4-MP1106融合蛋白的纯化制备 M:protein ladder;1:空BL21(DE3);2:诱导前全菌;3:诱导后全菌;4:细菌破碎后可溶部分;5:细菌破碎后沉淀部分;6:2% Triton X-100洗涤液;7:6 mol/L尿素变性上清;8:Ni2+-NTA流穿;9:50 mmol/L咪唑洗涤;10:500 mmol/L咪唑洗脱;11:对pH4缓冲液透析复性 |
选用2%的Triton X-100溶液洗涤DAMP4-MP1106融合蛋白包涵体,结果如图 4泳道6所示,部分杂蛋白被洗涤除去。
使用尿素浓度依次为3-8 mol/L的变性缓冲液分别溶解等量的DAMP4-MP1106融合蛋白包涵体,SDS-PAGE电泳检测溶解上清,结果如图 5所示。随着变性剂尿素浓度增高,对包涵体蛋白的溶解能力也逐渐提高,其中溶解杂蛋白的能力在尿素浓度6 mol/L和7 mol/L间有大幅度地提高。选用含有6 mol/L尿素的缓冲液溶解DAMP4-MP1106融合蛋白包涵体,进行后续纯化。如图 4泳道7所示,DAMP4-MP1106融合蛋白纯度提高到54.2%。

|
| 图 5 使用梯度浓度尿素变性溶解DAMP4-MP1106融合蛋白包涵体 3-8:溶解DAMP4-MP1106融合蛋白包涵体的溶液所含尿素浓度(mol·L-1) |
按照Ni2+-NTA亲和层析方法对DAMP4-MP1106融合蛋白进行纯化,图 4泳道8为未结合的杂蛋白流穿,图 4泳道9为低浓度咪唑(50 mmol/L)洗涤下来的结合不牢固或非特异性结合的杂蛋白,图 4泳道10为高浓度咪唑(500 mmol/L)洗脱下来的目的蛋白。纯化后,DAMP4-MP1106融合蛋白纯度达到了90.7%。
2.4 DAMP4-MP1106融合蛋白复兴条件筛选将DAMP4-MP1106融合蛋白的6 mol/L尿素溶液使用pH1-12的缓冲液和超纯水进行10倍稀释,测定其A310值。结果如图 6所示,pH1-5的A310值较低,对应的DAMP4-MP1106融合蛋白的溶解度较高;pH5到pH6间A310值迅速升高,对应着DAMP4-MP1106融合蛋白的溶解度迅速降低;超纯水溶液所对应的DAMP4-MP1106融合蛋白的溶解度也较低。鉴于此,本研究选用pH4的缓冲液透析DAMP4-MP1106融合蛋白进行蛋白复性,结果如图 4泳道11所示,软件检测分析,DAMP4-MP1106融合蛋白纯度提高到94.7%。

|
| 图 6 MP1106融合蛋白的溶解度与pH的关系 |
将纯化制备得到的水溶性DAMP4-MP1106融合蛋白使用TEV蛋白酶酶切,结果如图 7所示,泳道1为TEV蛋白酶,分子量约25 kD。泳道2为TEV蛋白酶室温酶切2 h后的DAMP4-MP1106融合蛋白样品,可清晰的看到DAMP4标签蛋白(Mw约15.6 kD)和抗菌肽MP1106(Mw约4.4 kD),切割比较完全,并通过二次Ni2+-NTA亲和层析纯化制备抗菌肽MP1106,纯度为99%。

|
| 图 7 TEV蛋白酶切DAMP4-MP1106融合蛋白 M:protein ladder;1:TEV蛋白酶;2:蛋白酶TEV酶切His6-DAMP4-TEV-MP106融合蛋白 |
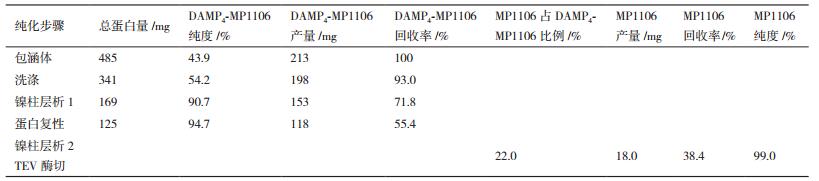
对整个纯化过程中蛋白的纯化和收率做了总结(表 1),实验过程中均采用BCA蛋白定量试剂盒测定每个样品的蛋白含量,采用Bio-Rad凝胶成像仪Quantify One软件检测SDS-PAGE电泳图,测定目的蛋白的纯度。经纯化复性,1 L培养物可以纯化得到18 mg纯度约99%的抗菌肽MP1106,按照抗菌肽MP1106的理论总产量,收率为38.4%。
MP1106分子内含有3对二硫键,使用含或不含20 mmol/L DTT的SDS-PAGE电泳制样缓冲液对DAMP4-MP1106融合蛋白和抗菌肽MP1106进行电泳检测对比分析,结果如图 8所示。泳道1(未加入20 mmol/L DTT的DAMP4-MP1106融合蛋白)和泳道2(加入20 mmol/L DTT的DAMP4-MP1106融合蛋白)相比,20 mmol/L DTT仅引起DAMP4-MP1106融合蛋白电泳条带稍微向上跃迁,差异不明显。泳道3(未加入20 mmol/L DTT的MP1106) 和泳道4(加入20 mmol/L DTT的MP1106) 相比,20 mmol/L DTT引起MP1106电泳条带明显向上跃迁。

|
| 图 8 MP1106分子内二硫键鉴定电泳图 1:不含有20 mmol/L DTT的DAMP4-MP1106融合蛋白样品;2:含有20 mmol/L DTT的DAMP4-MP1106融合蛋白样品;3:不含有20 mmol/L DTT的MP1106样品;4:含有20 mmol/L DTT的MP1106样品 |
前期文献报道,DAMP4标签蛋白具有在高盐缓冲液中耐高温的性能,已被开发成通用的小分子量蛋白或短肽的纯化工具[17-19]。本研究按照文献报道的方法,使用高盐缓冲液悬浮菌体,并进行加热处理,离心分离上清和沉淀,SDS-PAGE电泳检测分析显示,DAMP4-MP1106融合蛋白和其它杂蛋白在加热过程中全部变性沉淀,未达到纯化效果。经分析推测,可能由于抗菌肽MP1106短肽对DAMP4结构域产生了影响,造成包涵体表达。
菌丝霉素的空间结构由3个结构域组成(N-末端Loop环、两亲性α-螺旋中心、C-末端为两条反向平行的β-折叠),形成典型的CSαβ序。其中α-螺旋通过两对二硫键与β-折叠的一条链稳定相连,而N-末端的Loop环则通过一对二硫键与β-片层的另一条链相连[5, 20]。二硫键对蛋白质分子高级结构的稳定和生物学功能至关重要。有研究表明,蛋白分子内二硫键的形成,能使蛋白质分子的结构更加紧凑严密,表观分子量变小,SDS-PAGE电泳条带向下跃迁[21, 22]。本研究中加入20 mmol/L DTT后,DAMP4-MP106和MP1106电泳条带均有上移迹象,初步证明了MP1106分子内完成了二硫键的正确折叠。由于MP1106相对DAMP4分子量较小,故DAMP4-MP1106使用20 mmol/L DTT处理后,相对于MP1106电泳迁移率变化较小。
随着生物技术的飞速发展,利用微生物重组制备抗菌肽成为一种经济高效的方法。科学家们将菌丝霉素及其衍生物与蛋白标签(TrxA、GST、His6、SUMO和Intein等)基因融合,并在中间预设蛋白酶识别位点,采用大肠杆菌表达系统重组表达,纯化后经工具蛋白酶(Thrombin、Enterokinase、Factor Xa、SUMO和TEV等)酶切,获得具有生物功能活性的菌丝霉素,但其发酵产量较低、蛋白酶切效果差、回收率低[14-16]。国内Wang课题组[9, 11-13]利用毕赤酵母表达系统直接表达菌丝霉素及其衍生物多肽,发酵产量提高到克级每升,为其产业化奠定了坚实基础。但仍面临着一个世界性难题:菌丝霉素由于分子量小,易降解,体内半衰期短,药效持久性差,抗菌活性有待进一步提高等,至今尚无产品入市[9, 11-13]。本研究利用大肠杆菌系统表达DAMP4-MP1106融合蛋白,经蛋白复性、TEV酶切获得纯度为99%的抗菌肽MP1106 18 mg/L,酶切完全,回收率达到了38.4%,和已有研究报道相比,产量和回收率均得到了提高。菌丝霉素作为最具有应用前景的三大抗菌肽之一,作用机理清晰,对人体细胞无毒害作用[3],将菌丝霉素应用到临床,还需对其进行多方面功能性改造进化。所以,建立一个高效、快捷的菌丝霉素大肠杆菌表达系统对其功能性改造是至关必要的。
4 结论实现了融合蛋白DAMP4-MP1106在大肠杆菌中的表达,并经蛋白复性、纯化成功得到了目的蛋白抗菌肽MP1106。融合蛋白DAMP4-MP1106摇瓶发酵产量为118 mg/L,纯度为94.7%。经蛋白酶TEV酶切和二次Ni2+-NTA亲和层析,可获得纯度为99%的抗菌肽MP1106 18 mg/L,回收率达到了38.4%。通过简捷快速鉴定蛋白分子内的二硫键,初步证实了抗菌肽MP1106完成了分子内结构正确折叠。
| [1] | Mygind PH, Fischer RL, Schnorr KM, et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus[J]. Nature, 2005, 437 (7061): 975–980. DOI:10.1038/nature04051 |
| [2] | Czaplewski L, Bax R, Clokie M, et al. Alternatives to antibiotics-a pipeline portfolio review[J]. Lancet Infectious Diseases, 2016, 16 (2): 239–251. DOI:10.1016/S1473-3099(15)00466-1 |
| [3] | Hara S, Mukae H, Sakamoto N, et al. Plectasin has antibacterial activity and no affect on cell viability or IL-8 production[J]. Biochemical & Biophysical Research Communications, 2008, 374 (4): 709–713. |
| [4] | Harder, Jürgen. Antimicrobial Peptides Role in Human Health and Disease[M]. Bonn: Spriner, 2016. |
| [5] | Schneider T, Kruse T, Wimmer R, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor lipid Ⅱ[J]. Science, 2010, 328 (5982): 1168–1172. DOI:10.1126/science.1185723 |
| [6] | 张琪, 胡又佳, 陈习平, 等. 基于易错PCR的菌丝霉素的定向进化[J]. 中国医药工业杂志, 2014, 45(6). |
| [7] | Cao X, Yong Z, Mao R, et al. Design and recombination expression of a novel plectasin-derived peptide MP1106 and its properties against Staphylococcus aureus[J]. Applied Microbiology and Biotechnology, 2015, 99 (6): 2649–2662. DOI:10.1007/s00253-014-6077-9 |
| [8] | Lv LH, Luo XG, Ni M, et al. Indipendent and Tandem Expression of a novel antimicrobial peptides plectasin in Escherichia coli[C]. International Conference on Future Biomedical Information Engineering, 2011:134-138. |
| [9] | Zhang Y, Teng D, Mao R, et al. High expression of a plectasin-der-ived peptide NZ2114 in Pichia pastoris, and its pharmacodynamics, postantibiotic and synergy against Staphylococcus aureus[J]. Applied Microbiology and Biotechnology, 2014, 98 (2): 681–694. DOI:10.1007/s00253-013-4881-2 |
| [10] | TorresMK, DraghiDC, PillarCM, 等. Activity of NZ2114 against Staphylococcal and Streptococcal isolates, including resistant Phenotypes[J]. 中国人口资源与环境:英文版, 2008, 7(1): 30–36. |
| [11] | Yang Y, Teng D, Zhang J, et al. Characterization of recombinant plectasin:Solubility, antimicrobial activity and factors that affect its activity[J]. Process Biochemistry, 2011, 46 (5): 1050–1055. DOI:10.1016/j.procbio.2011.01.018 |
| [12] | Mao R, Da T, Wang X, et al. Design, expression, and characteriza-tion of a novel targeted plectasin against methicillin-resistant Stap-hylococcus aureus[J]. Applied Microbiology and Biotechnology, 2013, 97 (9): 3991–4002. DOI:10.1007/s00253-012-4508-z |
| [13] | Mao R, Teng D, Wang X, et al. Optimization of expression conditions for a novel NZ2114-derived antimicrobial peptide-MP1102 under the control of the GAP promoter in Pichia pastoris X-33[J]. BMC Microbiology, 2015, 15 (1): 389. |
| [14] | Jing XL, Luo XG, Tian WJ, et al. High-level expression of the antimicrobial peptide plectasin in Escherichia coli[J]. Current Microbiology, 2010, 61 (3): 197–202. DOI:10.1007/s00284-010-9596-3 |
| [15] | Chen X, Shi J, Chen R, et al. Molecular chaperones(TrxA, SUMO, Intein, and GST)mediating expression, purification, and antimicrobial activity assays of plectasin in Escherichia coli[J]. Biotechnology & Applied Biochemistry, 2015, 62 (5): 606–614. |
| [16] | Chen X, Wen YA, Li L, et al. The stability, and efficacy against Penicillin-resistant Enterococcus faecium, of the plectasin peptide efficiently produced by Escherichia coli[J]. Journal of Microbiology & Biotechnology, 2015, 25 (7). |
| [17] | Dwyer MD, Brech M, Yu L, et al. Intensified expression and purification of a recombinant biosurfactant protein[J]. Chemical Engineering Science, 2014, 105 (8): 12–21. |
| [18] | Dexter AF, Malcolm AS, Middelberg APJ. Reversible active switching of the mechanical properties of a peptide film at a fluid-fluid interface[J]. Nature Materials, 2006, 5 (6): 502. DOI:10.1038/nmat1653 |
| [19] | Middelberg AP, Dimitrijev-Dwyer M. A Designed biosurfactant protein for switchable foam control[J]. Chemphyschem, 2011, 12 (8): 1426. DOI:10.1002/cphc.201100082 |
| [20] | Mandal K, Pentelute BL, Tereshko V, et al. Racemic crystallography of synthetic protein enantiomers used to determine the X-ray structure of plectasin by direct methods[J]. Protein Science, 2009, 18 (6): 1146–1154. DOI:10.1002/pro.v18:6 |
| [21] | Wu L, Zhai Y, Lu J, et al. Expression, purification and preliminary characterization of glucagon receptor extracellular domain[J]. Protein Expression & Purification, 2013, 89 (2): 232. |
| [22] | Rogers MS, Baron AJ, McPherson MJ, et al. Galactose oxidase pro-sequence cleavage and cofactor assembly are self-processing reactions[J]. Journal of the American Chemical Society, 2000, 122 (5): 990–991. DOI:10.1021/ja993385y |