




2. 中国科学院系统微生物工程重点实验室, 天津 300308;
3. 中国科学院天津工业生物技术研究所, 天津 300308
2. Key Laboratory of Systems Microbial Biotechnology, Chinese Academy of Sciences, Tianjin 300308;
3. Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308
大肠杆菌(Escherichia coli)是常用的工业平台菌株, 其遗传改造工具成熟、遗传背景清楚, 具有操作简单、繁殖迅速和易培养等优点, 在生产过程中主要以葡萄糖、蔗糖和果糖为碳源, 生产效益受季节变化、天气状况以及农产品限制的影响。甲醇是一种重要的有机化工原料, 价格低廉, 来源广泛, 碳还原程度高, 是生物化工中糖质原料的理想替代品[1]。但E. coli等工业菌株不能利用甲醇作为碳源[2], 这限制了甲醇在生物化工领域的应用。近年来, 随着甲醇工业产能过剩的情况日益严重, 以及微生物利用甲醇机制研究的不断深入, 将甲醇利用途径导入E. coli等工业菌株并赋予其甲醇利用能力成为可能[3]。
核酮糖单磷酸(Ribulose monophosphate, RuMP)途径是目前研究较多的甲醇同化途径之一, 也是构建人工甲醇利用菌最常用的途径。RuMP途径广泛存在于甲基营养菌[4]中, 在甲醛浓度极低的情况下仍可高效捕获游离的甲醛[5]。在RuMP途径(图 1)中, 甲醇首先被甲醇脱氢酶(Methanol dehydrogenase, MDH)氧化生成甲醛, 甲醛和核酮糖-5-磷酸(Ribulose-5-phosphate, Ru5P)在3-己酮糖-6-磷酸合成酶(Hexulose-6-phosphate synthase, HPS)作用下缩合产生己酮糖-6-磷酸(Hexose-6-phosphate, H6P);H6P在6-磷酸-3-己酮糖异构酶(6-phospho-3-Hexuloisomerase, PHI)作用下异构化生成果糖-6-磷酸(Fructose-6-phosphate, F6P)。F6P在磷酸果糖激酶(Phosphate fructose kinase, PFK)催化下生成果糖-1, 6-二磷酸(Fructose-1, 6-diphosphate, F16dP), F16dP随后断裂形成甘油醛-3-磷酸(Glyceraldehyde-3-phosphate, G3P)和二羟基丙酮磷酸(Dihydroxyacetone phosphate, DHAP), 从而进入糖酵解[6]。同时, 一部分F6P通过一系列反应再生甲醛同化的受体Ru5P[7]。由于RuMP途径的所有反应均是放能的, 所以具有较高的甲醛同化效率[8]。

|
| 图 1 甲基芽孢杆菌(Bacillus methanolicus)代谢甲醇的RuMP途径 Ru5P:核酮糖-5-磷酸;R5P:核糖-5-磷酸;H6P:己酮糖-6-磷酸;X5P:木酮糖-5-磷酸;F6P:果糖-6-磷酸;F16dP:果糖-1, 6-二磷酸;G3P:甘油醛-3-磷酸;E4P:赤藓糖-4-磷酸;DHAP:二羟基丙酮磷酸;S17dP:景天庚酮糖-1, 7-二磷酸;S7P:景天庚酮糖-7-磷酸;MDH:甲醇脱氢酶;HPS:3-己酮糖-6-磷酸合成酶;PHI:6-磷酸-3-己酮糖异构酶;PFK:磷酸果糖激酶;FBA:果糖-二磷酸醛缩酶;RPI:核糖-5-磷酸异构酶;RPE:核酮糖-5-磷酸-3-差向异构酶;TKT:转酮醇酶;GLPX:果糖二磷酸酶/景天庚酮糖二磷酸酶 |
为了赋予常用工业菌株利用甲醇的能力, 多项研究试图利用RuMP途径改造平台菌株, Müller[2]和Witthoff[9]等分别在E. coli和谷氨酸棒杆菌(Corynebacterium glutamicum)中测试不同来源的MDH、HPS和PHI, 择优组装甲醇利用途径, 利用戊糖磷酸途径再生甲醛受体Ru5P。两组研究均使用了13C标记的甲醇作为底物, 通过胞内代谢物质谱分析, 证明了基因工程菌株在休止细胞状态下或利用葡萄糖生长时, 可同化少量的甲醇。Whitaker等[10]在E. coli中开展了利用甲醇合成柚皮素的研究, 通过使用酶活更高的MDH提高菌株利用甲醇的能力, 但是仍需添加其他碳源, 且13C标记的代谢物分析表明柚皮素中仅有3.5%的碳来自甲醇, 甲醇利用效率仍非常低。目前研究表明, 仅将RuMP途径导入工业菌株所构建的基因工程菌不能利用甲醇作为唯一碳源进行生长。这可能是由于甲醇利用途径关键酶的活性较低[11, 12], 也可能是异源途径与宿主内生代谢途径不匹配造成的[7, 13]。目前对甲醇利用限制因素的解析仍不清晰, 研究者难以开展针对性的遗传改造从而提高甲醇利用效率。
因此, 采用定向进化的方法, 对人工甲醇利用菌株进行进化, 可能是提高其甲醇利用效率的有效手段。基因组复制工程辅助的连续进化(Genome replication engineering assisted continuous evolution, GREACE)[14]技术是一种利用基因组复制协助连续进化来针对性提高微生物某种表型的手段。GREACE技术采用突变伴随筛选的方法, 利用修复矫正功能缺失的DNA聚合酶复合体(dnaQ基因突变体编码), 降低基因组复制过程中错配修复率, 加大突变率, 并有针对性的进行突变株筛选。GREACE技术已成功将E. coli对丁醇耐受性提高100倍, 对醋酸耐受性提高8倍[14], 是针对微生物耐受性进化的有效方法。
本研究在E. coli中组合MDH、HPS与PHI构建RuMP途径, 结合GREACE技术针对工程菌株外源导入基因与基因组进行定向进化, 通过甲醇为唯一碳源的压力筛选得到可以利用甲醇生长的突变株, 将突变株与原始菌株进行生长验证与13C标记示踪分析, 确定甲醇进入突变株代谢流并合成细胞碳骨架, 旨在为工业微生物提高甲醇利用能力提供有效的解决方案。
1 材料与方法 1.1 材料 1.1.1 主要试剂研究工作中所使用的E. coli质粒提取试剂盒及DNA胶回收试剂盒购自TIANGEN公司、基因合成由金唯智公司完成;酵母粉和胰蛋白胨购自英国Oxoid公司;琼脂粉、甘氨酸、异丙基-β-d-硫代吡喃半乳糖苷(Isopropyl-β-d-thiogalactopyranoside, IPTG)、卡那霉素(Kanamycin, Kan)、氨苄霉素(Ampicillin, Amp)、氯霉素(Chloroamphenicol, Cm)购自Solarbio公司;其他生化试剂均购自国药集团化学试剂有限公司。
1.1.2 培养基LB液体培养基:10 g/L蛋白胨、10 g/L NaCl、5 g/L酵母粉, pH7.0;M9液体培养基:17.1 g/L Na2HPO4·12H2O、3 g/L KH2PO4、0.5 g/L NaCl、1.0 g/L NH4Cl、0.24 g/L MgSO4与0.011 g/L CaCl2, pH7.0;M9辅助培养基:17.1 g/L Na2HPO4·12H2O、3 g/L KH2PO4、0.5 g/L NaCl、1.0 g/L NH4Cl、0.24 g/L MgSO4与0.011 g/L CaCl2、1 g/L酵母粉, pH 7.0。
1.1.3 质粒与菌株本研究所用的质粒与菌株由表 1所示。

将pZWM1质粒转化到E. coli BW25113ΔfrmA中, 将转化菌株株过夜活化后, 以1%的接种量将含有pZWM1质粒的E. coli BW25113ΔfrmA接种于30 mL LB液体培养基(100 mg/L Amp、50 mg/L Kan)中, 37℃ 220 r/min震荡培养至OD600约为0.6, 加入1 mmol/L IPTG诱导mdh3、hps和phi基因表达, 37℃ 220 r/min培养5 h进行诱导表达。将30 mL菌液高速离心弃上清, 菌体用3 mL磷酸盐缓冲液重悬, 冰浴条件下进行超声破碎细胞, 破碎条件为:功率40%, 工作1 s停3 s, 破碎时间15 min。破碎后溶液于4℃ 12 000 r/min离心10 min, 上清液即为可溶性的重组酶液, 加入4×NuPAGE® LDS Sample Buffer(Thermo Fisher公司)充分混合, 沸水浴15 min, 利用NuPAGE 4%-12% Bis-Tris预制胶(Thermo Fisher公司)进行SDS-PAGE电泳检测。
1.2.2 酶活测定 1.2.2.1 MDH酶活测定MDH活性检测方法参照文献[2], MDH催化甲醇的氧化反应伴随着烟酰胺腺嘌呤二核苷酸(Nicotinamide adenine dinucleotide, NADH)的生成, 因此可以通过检测NADH的生成检测MDH酶活。酶活测定方法为:将100 µL反应体系(含有0.5 mmol/L NAD+、1 mol/L甲醇、5 mmol/L MgSO4和50 mmol/L pH7.4的磷酸盐缓冲液)在37℃下孵育10 min, 加入37℃预热的100 µL粗酶液启动反应, 340 nm分光光度计下测定催化反应引起的NADH的增加。一个MDH酶活力(U)定义为:在37℃反应条件下, 每分钟将1 µmol NAD+还原为NADH所需的酶量。每个反应3个平行样, 取平均值作为最终酶活值, 对照反应为在相同条件下加入等量空质粒菌株的酶液。MDH比酶活力按照公式1计算, 蛋白浓度根据PierceTM BCA Protein Assay Kit(Thermo Fisher公司)测定。

HPS与PHI活性检测方法参照文献[9], HPS催化甲醛与Ru5P生成H6P, 后者被PHI催化生成F6P, 磷酸葡萄糖异构酶催化F6P生成葡萄糖-6-磷酸(Glucose 6-phosphate, G6P), G6P被磷酸葡萄糖脱氢酶催化生成葡萄糖酸-6-磷酸同时生成烟酰胺腺嘌呤二核苷酸磷酸(Nicotinamide adenine dinucleotide phosphate, NADPH), 通过检测NADPH的生成检测HPS与PHI的酶活力[9]。酶活测定方法为:将100 µL反应体系(5 mmol/L甲醛、5 mmol/L MgCl2、2.5 mmol/L NADP+、5 mmol/L R5P、2 U磷酸核糖异构酶、5 U磷酸葡萄糖脱氢酶、5 U磷酸葡萄糖异构酶、50 mmol/L pH 7.4的磷酸盐缓冲液)在37℃下孵育15 min, 使R5P与磷酸核糖异构酶充分反应生成底物Ru5P, 加入37℃预热的100 µL粗酶液启动反应, 340 nm分光光度计下测定催化反应引起的NADPH的增加。一个HPS-PHI(U)定义为:在37℃反应条件下, 每分钟将1 µmol NADP+还原为NADPH所需的酶量。每个反应均3个平行样, 取平均值作为最终酶活值, 对照反应为在相同条件下加入等量空质粒菌株的酶液。HPS-PHI偶联比酶活力按照公式2计算, 蛋白浓度根据PierceTM BCA Protein Assay Kit(Thermo Fisher公司)测定。

菌株25113ΔfrmA/pZWM1 & pKD46-Cm-dnaQ接入5 mL LB培养基中, 30℃ 220 r/min培养12 h, 将培养物作为种子接入20 mL LB传代培养基(100 mg/L Amp、50 mg/L Kan、10 mg/L Cm、1 g/L阿拉伯糖)中, 阿拉伯糖诱导dnaQ基因表达进行第一轮突变, 初始OD600 0.05, 30℃ 220 r/min培养12 h。取2 mL培养好的菌液接入20 mL LB传代培养基中进行第2轮突变, 30℃ 220 r/min培养12 h, 将剩余菌液用新鲜的M9培养基洗涤后转接到20 mL M9诱导培养基(5 g/L葡萄糖、50 mg/L Amp、0.5 mmol/L IPTG、20 mmol/L核糖)中, 其中IPTG诱导RuMP途径关键酶表达, 核糖为RuMP途径底物Ru5P提供前体, 初始OD600 0.6, 37℃ 220 r/min培养5 h。培养后菌液用新鲜M9洗涤后转接到100 mL M9筛选培养基(0.1 mmol/L IPTG、10 g/L甲醇)中筛选有益突变株, 检测初始OD600, 每24 h检测OD600。后续传代步骤筛选步骤与第二轮相同, 筛选到有明显生长的菌株进行分纯培养, 步骤与第二轮菌株筛选步骤相同。
1.2.4 甲醇为辅助碳源生长验证将菌株25113ΔfrmA/pTrc99A、25113ΔfrmA/pZWM1、25113ΔfrmA/pZWM1-13、25113ΔfrmA/pZWM1-14接入5 mL LB培养基中, 30℃ 220 r/min培养12 h, 将培养物作为种子接入M9辅助培养基(10 g/L甲醇、50 mg/L Amp、1 mmol/L IPTG)中, 初始OD600 0.05, 37℃ 220 r/min培养, 每3 h检测OD600。
1.2.5 13C标记代谢物培养菌株25113ΔfrmA/pZWM1-13按照定向进化第二轮诱导筛选步骤进行培养2 d, 第3天将菌体倒入50 mL离心管中, 8 000 r/min离心10 min。去上清液, 菌体用1.5 mL 6 mol/L HCl重悬并转移到有胶圈的气相瓶中, 100℃水解蛋白质24 h, 反应结束后20 000 r/min离心8 min, 上清液利用真空离心浓缩仪(Eppendorf公司)在60℃下干燥6-8 h。干燥样品溶于20 µL 50%乙腈, 快速震荡混匀后12 000 r/min离心10 min, 取上清液进行液相质谱检测检测。
1.2.6 13C标记代谢物分析检测系统由液相色谱(Shimdzu LC-30A)和质谱仪(Triple TOF 5600) 串联组成, 液相系统条件如下:色谱柱为SeQuant ZIC-HILIC(100 mm×2.1 mm, 3.5 μm粒径, 德国默克), A相:10 mmol/L醋酸铵、10 mmol/L氢氧化铵、5%乙腈;B相:100%乙腈, 液相色谱程序:0 min 3 min, 95% B;3 min 6 min, 95%-60% B;6 min 25 min, 60%-50% B;25 min-30 min, 50% B;30 min 30.5 min;50%-95% B;30.5 min 38 min, 95% B;色谱柱重新平衡10 min。流速为0.3 mL/min, 柱温为常温(25℃), 进样量为5 µL。本法使用Triple TOF 5600串联质谱仪, 采用电喷雾离子源(ESI), 负离子检测模式。质谱主要参数如下:雾化气(GS)55, 气帘气(CUR)35, 喷雾电压(ISVF)-4 500 V, 离子源温度(TEM)600℃。液相质谱结果利用软件PeakView进行分析, 根据氨基酸同位素峰面积进行积分, 13C标记比例按照公式3进行计算, 其中mi-1为氨基酸中(i-1) 个碳被13C标记占全部碳原子的比例, i为氨基酸碳原子总数。计算得到比例参照文献[17]进行除噪分析。
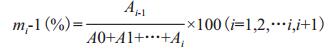
菌株25113ΔfrmA/pZWM1携带甲醇利用途径的相关基因mdh、hps和phi, 3个基因都受IPTG诱导型启动子Ptrc的控制。基因mdh全长1 158 bp, MDH分子量约为42 kD;基因hps全长636 bp, HPS分子量约为23 kD;基因phi全长564 bp, PHI分子量约为20 kD。重组菌株经IPTG在37℃诱导5 h后粗酶液SDS-PAGE, 结果如图 2所示, 与携带空质粒的对照菌株25113ΔfrmA/pTrc99A相比, 菌株25113ΔfrmA/pZWM1的粗酶液在40-50 kD和25 kD处有明显蛋白条带, 表明MDH、HPS和PHI均为可溶性蛋白, 但可能由于HPS与PHI分子量较为接近, SDS-PAGE无法区分两个蛋白。

|
| 图 2 SDS-PAGE检测MDH、HPS和PHI表达水平 M:Marker;1:25113ΔfrmA/pTrc99A粗酶液上清液;2:25113ΔfrmA/pZWM1粗酶液上清液 |
为检测外源表达的MDH、HPS和PHI的酶活, 对菌株25113ΔfrmA/pZWM1粗酶液的MDH、HPS和PHI酶活进行测定。重组菌株25113ΔfrmA/pZWM1粗酶液中MDH比酶活为0.64±0.038 mU/mg, HPS-PHI偶联比酶活为38.99±2.77 mU/mg。
2.3 重组菌株定向进化及筛选为获得有效利用甲醇进行生长的菌株, 利用GREACE技术对菌株25113ΔfrmA/pZWM1进行定向进化, 将携带dnaQ突变体的pKD46-Cm-dnaQ质粒转化到25113ΔfrmA/pZWM1中, 获得菌株25113ΔfrmA/pZWM1 & pKD46-Cm-dnaQ。进化过程中产生的有益突变株, 会使培养物中菌体量有所提高。5到13轮传代培养结果如图 3所示, ΔOD600为在筛选培养基中培养2 d后培养物OD600与初始OD600之差, 第5轮与第6轮传代培养物培养2 d后菌体OD600均呈现不同程度的下降, 第6轮传代培养物培养2 d后菌体量下降5.3%, 表明初期工程菌不能在筛选培养基中生长;从第7轮传代培养后菌体OD600均呈现不同程度的上升, 第10轮传代培养物利用甲醇生长优势明显, 菌体量增加78.2%。继续传代, 细胞利用甲醇的生长减弱。因此从第10轮培养物中筛选利用甲醇生长的突变株。

|
| 图 3 25113ΔfrmA/pZWM1连续传代获得的培养物利用甲醇生长情况 ΔOD600为在筛选培养基中培养2 d后培养物OD600与初始OD600之差 |
菌株25113ΔfrmA/pZWM1第10轮培养物利用甲醇时有微弱生长现象, 对第10代培养物进行三区划线, 挑取15个单克隆进行利用甲醇的生长测试。如图 4所示, 15个突变株中, 13号突变株培养2 d后菌株OD600与原始接菌OD600之差为0.124, 菌体量增加55.3%;14号突变株培养2 d后菌株OD600与原始接菌OD600之差为0.14, 菌体量增加82.4%。结果表明, 13号和14号突变株较其它突变株有明显生长, 将这两株突变株分别命名为25113ΔfrmA/pZWM1-13和25113ΔfrmA/pZWM1-14。

|
| 图 4 25113ΔfrmA/pZWM1突变株利用甲醇生长情况 ΔOD600为在筛选培养基中培养2 d后培养物OD600与初始OD600之差 |
为进一步验证突变株利用甲醇的生长情况, 使用1 g/L酵母粉作为碳源, 并加入10 g/L甲醇作为辅助碳源培养突变株25113ΔfrmA/pZWM1-13和25113ΔfrmA/pZWM1-14, 将进化出发菌株25113ΔfrmA/pZWM1与携带空载质粒的菌株25113ΔfrmA/pTrc99A作为对照。如图 5所示, 重组菌株25113ΔfrmA/pZWM1生长到OD600为0.58后停滞生长, 对照菌株25113ΔfrmA/pTrc99A生长到OD600到0.64后停滞生长, 菌株25113ΔfrmA/pZWM1生长减弱的现象可能是由于大量表达外源基因造成的。突变株25113ΔfrmA/pZWM1-13生长到OD600为0.75后停滞生长, 较出发菌株OD600增加27.6%;突变株25113ΔfrmA/pZWM1-14生长到OD600为0.70后停滞生长, 较出发菌株OD600增加21.2%。通过生长曲线验证结果表明, 相较于携带空质粒的对照菌株25113ΔfrmA/pTrc99A和进化的出发菌株25113ΔfrmA/pZWM1, 突变株25113ΔfrmA/pZWM1-13和25113ΔfrmA/pZWM1-14在利用甲醇作为辅助碳源时可以生产更多的生物量, 说明进化获得的突变株利用甲醇的细胞生长得到提高。

|
| 图 5 25113ΔfrmA/pZWM1原始菌株与突变株利用甲醇作为辅助碳源的生长曲线 |
为进一步验证突变株的甲醇利用情况, 我们使用13C标记甲醇培养突变株25113ΔfrmA/pZWM1-13, 之后将菌体进行酸水解[18], 对所得到的氨基酸进行液相质谱分析。质谱结果除噪后分析同位素含量的差异, 结果如图 6所示。12C甲醇培养的菌株(图 6-A)与13C基础培养的菌株(图 6-B)氨基酸同位素分布有明显差异, 与12C甲醇培养菌株相比, 13C甲醇培养菌株丙氨酸(Alanine, Ala)13C标记含量增加0.702%, 天冬氨酸(Aspartic acid, Asp)13C标记含量增加1.132%, 谷氨酸(Glutamic acid, Glu)13C标记含量增加0.211%, 苯丙氨酸(Phenylalanine, Phe)13C标记含量增加0.379%, 组氨酸(Histidine, His)13C标记含量增加1.163%, 甘氨酸(Glycine, Gly)13C标记含量增加0.667%, 赖氨酸(Lysine, Lys)13C标记含量增加1.029%, 丝氨酸(Serine, Ser)13C标记含量增加1.743%, 酪氨酸(Tyrosine, Tyr)13C标记含量增加1.435%, 甲硫氨酸(Methionine, Met)13C标记含量增加7.236%, 缬氨酸(Valine, Val)13C标记含量增加1.359%。结果表明, 13C培养菌株氨基酸同位素比例与12C培养菌株相比有明显增加, 表明甲醇进入细胞代谢并参与细胞骨架合成。

|
| 图 6 突变株25113ΔfrmA/pZWM1-13代谢12C标记甲醇(A)与13C标记甲醇(B)时生物量中同位素含量分析 |
近年来, 越来越多的研究者认识到甲醇生物转化利用的重大意义, 对甲醇利用途径的研究也越来越深入[7, 19]。借助于已解析的甲醇利用途径和先进的代谢工程技术手段, 多个课题组采用RuMP途径对常用工业平台菌株进行了改造, 试图赋予其甲醇利用能力。通过异源表达mdh、hps和phi基因, 基因工程菌株可以利用少量的甲醇, 但距离利用甲醇作为主要碳源甚至唯一碳源生长还有很大距离[2, 9, 10, 20]。本研究以E. coli为底盘菌, 结合代谢工程与定向进化的技术手段, 在E. coli胞内构建RuMP途径的基础上开展GREACE进化, 获得了利用甲醇生长更优的突变株, 为下一步限制因素解析和菌种优化提供了新的研究对象和思路。
打通甲醇利用途径是构建高效人工甲醇利用菌株的第一步, 要提高工程菌株的甲醇利用效率, 需要有明确的靶点, 但是目前对甲醇利用限制因素的了解还非常少[7]。在这种情况下, 采用定向进化的方式, 优先获得甲醇利用能力得到提高的菌株, 可以为解析甲醇利用的限制因素提供良好的模型, 指导人工甲醇利用菌株的改造和优化。GREACE进化通过引入dnaQ基因突变体, 使菌株DNA复制过程中的矫正功能缺陷, 通过连续传代不断在菌株外源基因与基因组上积累随机突变[14], 在此过程中, 每一代培养物均可能出现针对甲醇生长的有益突变和有害突变。针对导入RuMP途径的E. coli不能在甲醇为唯一碳源的培养基进行生长这一特点, 将传代培养物进行以甲醇为唯一碳源的压力筛选, 进一步对有益突变进行富集。在对菌株25113ΔfrmA/pZWM1进行GREACE进化过程中, 每一代培养物在利用甲醇时具有不同的细胞生长情况, 这可能是由于GREACE引入突变的随机性导致的。一些对细胞生长或甲醇代谢有害的突变, 可能会抑制细胞生长。第10代培养物利用甲醇时体现出最优的细胞生长, 在进行纯化时, 两株突变株(25113ΔfrmA/pZWM1-13和25113ΔfrmA/pZWM1-14) 具有明显的生长现象, 菌体量最高增加82.4%, 筛选得到的突变株能够更好的利用甲醇并合成细胞碳骨架, 因此GREACE进化并配合压力筛选的方法适用于工业菌株甲醇利用效率的提高。
利用甲醇作为辅助碳源时, 突变株25113ΔfrmA/pZWM1-13具有最好的细胞生长情况。为了对该突变株的甲醇利用情况进行表征, 使用13C标记甲醇作为底物培养突变株25113ΔfrmA/pZWM1-13, 收集菌体后进行酸水解, 并通过液相质谱分析酸水解产生的氨基酸中13C分布情况。Ala、Leu和Val以丙酮酸为前体, Asp和Lys以草酰乙酸为前体, Glu和Pro以α-酮戊二酸为前体, Phe、Tyr和Try以磷酸烯醇式丙酮酸为前体, Gly以3-磷酸甘油醛为前体, 因此菌体蛋白氨基酸的水平可以在一定程度上反映胞内代谢物的水平[18]。检测结果显示Ala、Asp、Glu、Phe、His、Gly、Lys、Ser、Tyr、Met和Val在突变株代谢13C标记甲醇时具有明显的同位素比例差异, 说明甲醇通过RuMP途径进入了中心代谢, 并经过EMP途径和TCA循环等途径生成了相应的胞内代谢物与氨基酸, 参与了菌体蛋白合成。对突变株进行深入的分析, 可能由此获得改造靶点, 为进一步提高菌株的甲醇利用能力奠定基础。
4 结论本研究在E. coli中异源表达RuMP途径关键酶MDH、HPS和PHI, 构建了甲醇同化途径。并采用GREACE进化的方法, 在细胞复制过程中引入随机突变, 结合以甲醇为碳源的压力筛选, 获得了利用甲醇生长较优的突变株, 13C标记的质谱分析表明突变株可以利用甲醇合成细胞碳骨架。为解析甲醇利用限制因素和甲醇利用菌株的优化提供了新的研究对象和思路。
致谢 衷心感谢李庆刚老师和张志丹老师在实验过程中提供的帮助, 特别感谢郑小梅老师对文章的修改提供宝贵意见。| [1] | Choo S, Um Y, Han SO, et al. Engineering of Corynebacterium glutamicum to utilize methyl acetate, a potential feedstock derived by carbonylation of methanol with CO[J]. Journal of Biotechnology, 2016, 224 : 47–50. DOI:10.1016/j.jbiotec.2016.03.011 |
| [2] | Muller JE, Meyer F, Litsanov B, et al. Engineering Escherichia coli for methanol conversion[J]. Metabolic engineering, 2015, 28 : 190–201. DOI:10.1016/j.ymben.2014.12.008 |
| [3] | Schrader J, Schilling M, Holtmann D, et al. Methanol-based indus-trial biotechnology: current status and future perspectives of meth-ylotrophic bacteria[J]. Trends in Biotechnology, 2009, 27 (2): 107–115. DOI:10.1016/j.tibtech.2008.10.009 |
| [4] | Kalyuzhnaya M, Puri AW, Lidstrom ME. Metabolic engineering in methanotrophic bacteria[J]. Metabolic Engineering, 2015, 29 : 142–152. DOI:10.1016/j.ymben.2015.03.010 |
| [5] | Kato N, Yurimoto H, Thauer RK. The physiological role of the ribulose monophosphate pathway in bacteria and archaea[J]. Bioscience Biotechnology & Biochemistry, 2006, 70 (1): 10–21. |
| [6] | 晁红军, 宋修鹏, 孙继华, 等. 甲基营养菌的研究进展[J]. 微生物学通报, 2009, 36(11): 1727–1737. |
| [7] | Whitaker WB, Sandoval NR, Bennett RK, et al. Synthetic methylotrophy: engineering the production of biofuels and chemicals based on the biology of aerobic methanol utilization[J]. Current Opinion in Biotechnology, 2015, 33 : 165–175. DOI:10.1016/j.copbio.2015.01.007 |
| [8] | 宋中邦, 陈丽梅, 李昆志, 等. 细菌的核酮糖单磷酸途径与甲醛同化作用[J]. 微生物学报, 2007, 47(1): 168–172. |
| [9] | Witthoff S, Schmitz K, Niedenfuhr S, et al. Metabolic engineering of Corynebacterium glutamicum for methanol metabolism[J]. Applied and environmental microbiology, 2015, 81 (6): 2215–2225. DOI:10.1128/AEM.03110-14 |
| [10] | Whitaker WB, Jones JA, Bennett K, et al. Engineering the biological conversion of methanol to specialty chemicals in Escherichia coli[J]. Metabolic Engineering, 2017, 39 : 49–59. DOI:10.1016/j.ymben.2016.10.015 |
| [11] | Ochsner AM, Muller JE, Mora CA, et al. In vitro activation of NAD-dependent alcohol dehydrogenases by Nudix hydrolases is more widespread than assumed[J]. Febs Letters, 2014, 588 (17): 2993–2999. DOI:10.1016/j.febslet.2014.06.008 |
| [12] | Wu TY, Chen CT, Liu TJ, et al. Characterization and evolution of an activator-independent methanol dehydrogenase from Cupriavidus necator N-1[J]. Applied Microbiology and Biotechnology, 2016, 100 (11): 1–15. |
| [13] | Price JV, Chen L, Whitaker WB, et al. Scaffoldless engineered enzyme assembly for enhanced methanol utilization[J]. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113 (45): 12691–12696. DOI:10.1073/pnas.1601797113 |
| [14] | Luan GD, Cai Z, Li Y, et al. Genome replication engineering assisted continuous evolution(GREACE)to improve microbial tolerance for biofuels production[J]. Biotechnology for Biofuels, 2013, 6 (1): 1–11. DOI:10.1186/1754-6834-6-1 |
| [15] | Amann E, Ochs B, Abel KJ. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli[J]. Gene, 1988, 69 (2): 301–15. DOI:10.1016/0378-1119(88)90440-4 |
| [16] | Ishikawa H, Yoshihara M, Baba A, et al. Formation of zinc protoporphyrin Ⅸ form myoglobin with pork loin extract[J]. Journal of the Faculty of Agriculture Kyushu University, 2006, 51 (1): 93–97. |
| [17] | Wahl SA, Dauner M, Wiechert W. New tools for mass isotopomer data evaluation in 13C flux analysis: Mass isotope correction, data consistency checking, and precursor relationships[J]. Biotechnology & Bioengineering, 2004, 85 (3): 259–68. |
| [18] | You L, Page L, Feng X, et al. Metabolic pathway confirmation and discovery through C-labeling of proteinogenic amino acids[J]. Journal of Visualized Experiments, 2012, 59 (59): e3583–3689. |
| [19] | Kalyuzhnaya MG, Puri AW, Lidstrom ME. Metabolic engineering in methanotrophic bacteria[J]. Metabolic Engineering, 2015, 29 : 142–152. DOI:10.1016/j.ymben.2015.03.010 |
| [20] | Leβmeier L, Pfeifenschneider J, Carnicer M, et al. Production of carbon-13-labeled cadaverine by engineered Corynebacterium glutamicum using carbon-13-labeled methanol as co-substrate[J]. Applied Microbiology and Biotechnology, 2015, 99 (23): 10163–76. DOI:10.1007/s00253-015-6906-5 |