




2. 中国农业大学食品科学与营养工程学院,北京 100083
2. College of Food Science and Nutritional Engineering, China Agricultural University, Beijing 100083
核酸体外扩增技术在分子生物和生物分析等领域是一项重要的技术,可用于定性、定量地分析和检测微量核酸,其在临床医学、检验医学、分子生物学、基因组学及食品安全等相关的各个领域发挥着重要的作用,是生命科学发展不可或缺的一种重要检验方法。随着生物技术在临床和现场检测方面要求的不断提升,越来越多的研究人员开始把目光专注于核酸体外扩增新技术的研究。聚合酶链式反应(Polymerase chain reaction,PCR)由于其特异性强,灵敏度高等优点,是目前应用最为广泛的核酸体外扩增技术。虽然PCR技术已经应用于分子生物学等各个领域,但是该技术需要通过不断的变化温度才能实现核酸扩增,因此,热循环仪是不可或缺的设备。然而,由于热循环仪体积大、价格较高,使其在基层的推广和现场检测受到了极大的限制[1]。因此,陆续出现了不同的等温扩增技术,这些技术反应温度单一,不受热循环仪的制约,正逐渐成为PCR的最佳替代方法。
20世纪90年代初,很多研究人员看到了突破传统核酸体外扩增技术将会带来核酸体外扩增的新革命,从而尝试创新发展无须热变性的核酸恒温扩增技术,并挖掘其在各领域应用的潜力,这些核酸等温扩增的新技术应用于生命科学及相关的各个领域是必然趋势,也吸引了越来越多研究者的目光。
等温扩增技术的基本原理和反应成分各不相同,依据等温扩增的基本原理和步骤,本文将现有等温扩增总结为两大类,第一类是第一步反应依赖于特异性引物延伸的等温扩增技术;第二类是第一步反应依赖于限制性内切酶的等温扩增技术。本文概述并比较分析了核酸等温扩增技术的优缺点及反应原理等,以期对相关领域的研究起到积极的促进作用。
1 依赖于特异性引物延伸的等温扩增技术与传统PCR技术相似,为了保证目的片段特异性扩增,避免非特异产物的形成,在众多等温扩增方法中,依然使用特异性引物退火延伸作为等温扩增的起始步骤,如依赖核酸序列扩增技术、滚环扩增技术、环介导等温扩增等。但是,由于引物与目的模板退火结合的机理,这就不可避免地要对非单链的起始靶物质进行高温变性处理,这些方法并不是彻底的等温扩增。随着技术的发展,为了避免高温热变性步骤,DNA解旋酶、单链结合蛋白等被引入等温扩增体系。
1.1 依赖核酸序列扩增技术依赖核酸序列扩增技术(Nucleic acid sequence-based amplification,NASBA)于1991年由Compton等[2]首次提出,该方法结合了转录依赖扩增系统(Transcription-based amplification system,TAS)[3]和自主序列复制系统(Self-sustained sequence replication,3SR)2种方法[4],并对其进行改进。
NASBA技术与3SR相似,依赖AMV逆转录酶、RNase H和T7 RNA聚合酶和一对引物来完成等温扩增,整个反应包含非循环相和循环相。在对目的RNA片段进行扩增时,以指数扩增方式大量生成单链RNA[5]。不过,与3SR相比,NASBA使用了DMSO(二甲基亚砜,Dimethyl sulfoxide,DMSO),反应温度也适当地从37℃提高到41℃,有效地提高了反应的特异性[6]。随后,Gene-Probe公司基于NASBA成功研制出依赖转录介导扩增(Transcription mediated amplification,TMA)检测试剂盒[7],带有RNase H活性和逆转录酶活性的MMLV逆转录酶(鼠白血病逆转录酶)取代了AMV逆转录酶、RNase H。由于3SR,NASBA和TMA的原理十分相似,所以较多文献将这3种技术或其中两种视为同一种[5, 7-9]。
NASBA除了应用于RNA扩增,经过适当的改良也可以扩增DNA[6]:一种方法是使用限制性内切酶切割双链DNA,使其末端产生T7 RNA聚合酶启动子序列[2];另一种方法是在非循环相里进行两次95℃变性,第一次变性,双链DNA解离成单链DNA,引物退火结合上后,在AMV反转录酶的作用下,延伸形成新的双链DNA,接下来进行第二次变性,从而获得了5'端含有T7 RNA聚合酶启动子序列的单链DNA。
NASBA由于其快速高效、操作简便、设备要求低、扩增对象广等特点,已被广泛应用与发展。Lau等[10]分别使用NASBA-电化学发光法(NASBA-ECL)和NASBA-酶连接寡核苷酸捕获技术(NASBA-EOC)两种检测方法检测出口蹄疫病毒;Zhao等[11]通过微流控芯片实时免疫NASBA检测出水传染病原菌;Clancy等[12]开发出一种带有扩增内标的双重实时NASBA诊断方法,成功检测引起细菌性脑膜炎的流感嗜血杆菌,脑膜炎奈瑟氏菌和脑炎链球菌。Hønsvall等[13]使用实时NASBA技术扩增微小隐孢子虫和人隐孢子虫的MIC1转录本,成功区分以上两种隐孢子虫,在10 μL保存悬液中,检测灵敏度可低至5个隐孢子虫卵囊。Zeng等[14]结合NASBA和ELISA两种技术优势,开发出NASBA-ELISA检测方法,实现高效简便检测草鱼呼肠孤病毒。尽管NASBA存在众多优势,但是其缺点也不能被忽视:首先,其不是完全意义上的等温扩增技术,不能进行一步扩增;其次,酶的热稳定性差,不能在反应之前加入;最后,有效扩增片段的长度较短,仅100-250 bp之间[6]。
1.2 滚环扩增技术Fire等[15]于1995年首次提出滚环扩增技术(Rolling circle amplification,RCA),该技术是借鉴微生物环状DNA复制过程建立起来的一种体外等温扩增技术。在RCA反应中,phi29 DNA聚合酶是至关重要的组成部分,具有持续DNA合成能力和链置换活性[16, 17]。
RCA的反应模板通常是单链环状DNA,因此,须对双链DNA样品进行处理。如果样品是双链环状DNA,则变性处理即可;但如果样品是双链线状DNA,在进行变性处理后,还需用T4多核苷酸激酶与T4 DNA连接酶环化单链线状DNA[15]。RCA按引物数量可分为单引物RCA,双引物RCA和多引物RCA[18]。
1.2.1 单引物RCA只需要一条引物P1即可完成扩增。引物P1与单链环状DNA结合后,在phi29 DNA聚合酶的作用下开始延伸,当合成至自身5'端时,在phi29 DNA聚合酶链置换活性的作用下,5'端DNA开始被置换下来,并继续以环状DNA为模板进行线性合成,最终生成一条具有重复序列且与模板DNA完全互补的线状单链DNA。
1.2.2 双引物RCA也称为超分支滚环扩增[19],在单引物RCA的基础上,再增加一条与扩增产物互补的引物P2,P2可与单引物RCA的产物退火延伸。随着反应的进行,P2延伸链开始将下游P2的延伸链置换下来,被置换下来的P2延伸链作为模板与P1结合。同样地,上游P1延伸链会把下游P1延伸链置换下来,如此形成指数扩增,最终得到一系列长短不一的双链DNA产物。
1.2.3 多引物RCA[20]由phi29 DNA聚合酶、单链环状DNA模板和随机引物组成。多引物RCA原理与双引物RCA相似,随机引物可以在环状DNA的不同位置同时延伸产生多个复制叉,然后在聚合酶的作用下置换非模板链,在置换非模板链的同时,其他随机引物也开始与非模板链退火延伸,进而合成双链DNA[21]。与单引物RCA和双引物RCA相比,多引物RCA在合成速率和产量方面得到了明显提高[20]。
到目前为止,出现了许多原理与RCA相似或衍生于RCA的核酸扩增技术,如利用T7 RNA聚合酶建立的以单链环状DNA为模板的滚环扩增技术[22];用于扩增基因组DNA的多重置换扩增技术(Multiple displacement amplification,MDA)[23-25];利用锁式探针的信号扩增RCA[19]。在微生物检测方面,Rockett等[26]开发出依赖特异性引物的dRCA技术,在存在人源DNA的情况下,成功特异扩增BKV,HPyV6,HPyV7,TSPyV和STLPyV等7种人多瘤病毒,相比使用随机引物的传统RCA法,dRCA具有更强的灵敏度和更高的产量。Wen等[27]使用RCA技术扩增埃博拉病毒基因,同时基于氧化石墨烯吸附FAM的性质,开发出一款新型荧光生物传感器,成功对扩增产物进行检测,检出限低至1.4 pM,即使在1%人血清中,也可检测出目的基因。Hao等[28]基于RCA技术和WS2纳米片,发明了一种用于检测金黄色葡萄球菌的加强化学发光能量共振转移传感器,其检出限可达15 CFU/mL。RCA与其他核酸扩增方法相比,其主要优点为:反应机制简单,仅需一种聚合酶即可实现核酸扩增;灵敏度高,一条引物可达到105倍扩增[29],而两条引物的扩增效率可达到109倍[19];不需要PCR仪,避免了热循环过程对反应组分的影响。RCA的不足之处在于:前期处理繁琐,反应模板要求是单链环状DNA,须对不符合要求的样品进行退火和环化处理;反应时间较长,至少需要4 h[15, 20]。
1.3 环介导等温扩增环介导等温扩增(Loop-mediated isothermal amplification,LAMP)是Notomi等[30]于2000年开发的一种具有快速高效、特异性强、录敏度高等特点的等温扩增技术。LAMP的反应过程可分为3个阶段:循环模板合成阶段、循环扩增阶段和伸长再循环阶段[18, 30]。LAMP使用4条引物,分别是正向内引物FIP(包括F1c和F2两部分序列,其中c代表反向互补序列),反向内引物BIP(包括B1c和B2),正向外引物F3和反向外引物B3。
反应初始,FIP和F3先后与单链DNA模板结合,引发循环模板合成,随着F3的延伸,FIP产生的延伸链被置换出来,同时,其5'端发生自我碱基配对,形成环状结构。随即,BIP和B3以环状结构DNA作为模板延伸,引发新一轮链置换反应,最终形成一条哑铃状单链DNA,即循环模板。进入循环扩增阶段后,哑铃状单链DNA 3'端以自身为模板进行延伸,打开5'端环状结构,形成一条双链茎环DNA。随后,FIP的F2区域与茎环DNA环状结构中的F2c区域互补配对,发生延伸置换反应,自此引发循环扩增和伸长再循环扩增,进入指数扩增阶段。随着FIP与BIP不断与环状结构结合,链置换反应持续发生,最终大量生成不同长度地多环花椰菜结构DNA,而每条DNA则是由交替反向重复靶序列构成[5, 30]。LAMP原理虽然复杂,但是实际操作简单,是一种简单、快速、有效的核酸扩增技术。
LAMP的主要优点是特异性强,多条互补序列的引入使得即使存在非目的DNA的情况下,也不会对靶序列的扩增产生明显影响;其次,LAMP扩增效率高,由于存在多个循环体系,可实现高效扩增,其检测限可低至几个拷贝,远优于PCR[18, 30]。再次,LAMP扩增产物检测方便,LAMP会产生焦磷酸镁白色沉淀,通过肉眼或浊度仪即可判定体系是否发生DNA扩增,适合用于即时检测(Point-of-care testing,POTC)。
在10多年的时间里,不断有研究者从模板性质[31]、反应抑制因子[32]、环引物的引入[33]及多重检测[34-37]等方面对LAMP进行改进。到目前为止,由于引物设计的复杂性,仅发展到五重检测。由于LAMP反应成本低、扩增时间短,无论是实验室研究还是现场检测都可以准确、快速、灵敏的完成,所以在基层检验的推广中具有良好的前景,是真正普及型的核酸检测方法。科学家们已经将LAMP技术成功应用于病毒、致病菌及转基因作物的检测[38]。
尽管LAMP扩增技术应用十分广泛,但是不可避免的,LAMP也存在一定的缺陷:首先,由于LAMP产物鉴定结果只有扩增与不扩增,所以如果发生非特异性扩增,则不易识别。其次,其基本原理是基于链置换和链取代两种过程,LAMP的扩增片段长度一般在200-300 bp,限制了LAMP在扩增长片段方面的应用。另外,由于高灵敏度,操作过程中极易发生污染从而产生假阳性结果;LAMP在产物的回收、鉴定、克隆和单链分离等方面也存在很大的操作障碍。
1.4 依赖解旋酶扩增技术DNA体内复制主要依赖于DNA解旋酶、DNA聚合酶以及各种辅助因子,根据以上复制机制,Vincent等[39]模拟出依赖解旋酶扩增技术(Helicase-dependent amplification,HDA)。HDA是一种彻底的等温DNA体外扩增技术,目前,能够达到反应全过程恒温这一条件的体外等温扩增技术屈指可数,其全程等温的实现主要依赖于大肠杆菌UvrD解旋酶。
HDA的原理分为两个阶段:第一阶段,核酸解旋酶打开DNA双链,使目的模板链解链为单链状态;单链结合蛋白与之结合,它的作用是保证单链结构不发生复性;第二阶段,DNA聚合酶各自以单链为模板由引物为起始生成新链,新生成的双链DNA作为下一轮扩增的模板,如此反复循环扩增实现靶DNA量的指数增加。HDA的反应步骤与PCR相似,均包括双链DNA解链、退火及延伸等步骤,但最大的区别是采用酶维持单链作用替代升温解双链过程。
HDA的出现引起了许多研究者的关注,并对其进行了大量的研究及改进。An等[40]报道了嗜热HDA(Thermophilic helicase-dependent amplification,tHDA),该技术将原先使用的大肠杆菌UvrD换成了来自腾冲嗜热杆菌Thermoanaerobacter tengcongensis的耐热UvrD解旋酶(Tte-UvrD)。该酶的应用使反应可在60-65℃之间进行,温度的提升增强了该技术的灵敏度和特异性。Li等[41]在未提取DNA的前提下,直接使用tHDA成功检测出人体血液中的疟原虫,检出限达到50拷贝。Motré等[42]研发出了一种具有两种不同功能的新酶,既可以解旋双链DNA,又可以进行聚合反应,解决了解旋酶与聚合酶协作效率低的问题。这种酶实质上是Tte-UvrD和Bst DNA聚合酶通过卷曲螺旋域连接在一起的复合体。该新酶的使用明显提高了扩增片段的长度。除了前面提到的两种发展外,Xu等[43]的cHDA和Goldmeyer等[44]的RT-tHDA也对传统HDA进行了改良。
HDA作为新型等温核酸扩增技术的优势在于:反应全程温度恒定,无需调节温度,克服了传统PCR通过“变性-退火-延伸”过程的多次变温循环的瓶颈;反应机制简单,仅需两条简单的引物;反应时间较短,在1-1.5 h就可以实现扩增。然而HDA也存在其缺陷,目前,常用的解旋酶是大肠杆菌MvrD解旋酶,其解链速度为20 bp/s,解旋速度慢及持续能力差,因此HDA只适用于扩增短片段[39]。
1.5 重组酶聚合酶扩增继HDA之后,Piepenburg等[45]于2006年创建了一种新型的全程等温扩增技术,其与HDA相似,也使用酶来打开双链DNA,该技术称为重组酶聚合酶扩增(Recombinase polymerase amplification,RPA)。RPA是参照了T4噬菌体DNA复制系统,该反应系统中除了需要一种常温下能工作的DNA聚合酶外,还包含噬菌体μvsX重组酶和单链DNA结合酶gp32,目前有些实验中还会加入辅助μvsX重组酶的μvsY蛋白。
重组酶介导等温扩增(Recombinase aid amplification,RAA)是基于RPA的一种改良,最大的突破是重组酶的选择上。RPA使用的是噬菌体μvsX重组酶,而RAA使用的是从细菌或真菌中获得的重组酶。这类酶不仅来源更为广泛,而且最重要的是它在37℃恒温下便可与引物紧密结合形成酶-引物聚合体,反应的温度适应性更强[46]。
两种方法的扩增原理为:引物常温条件下与重组酶结合形成一种结构稳定的复合物,当引物在模板DNA上搜索到与之完全互补的序列时发生退火;退火发生的同时单链DNA结合蛋白被激发,发生作用使模板DNA保持单链状态,引物在DNA聚合酶催化作用下延伸形成新的DNA互补链。RAA和RPA反应迅速快捷,通常在1 h内即能得到可以用琼脂糖凝胶电泳检测到的扩增产量[47]。
与HDA相同,用于检测PCR产物的方法也可用于RPA。Piepenburg等设计了含有一个四氢呋喃(Tetrahydrofuran,THF)分子的荧光探针,并在THF两侧的核苷酸上分别标记一个报告荧光基团和一个淬灭荧光基团。同时,为了防止探针被作为引物延伸,对其3'端进行修饰。当探针结合到模板上时,来自大肠杆菌的核酸内切酶Ⅳ(Nfo)识别切割THF,上游探针形成新的3'端,在Bsu聚合酶的作用下,作为引物延伸,置换出下游探针,从而使两个荧光基团分离,产生荧光信号。为了实现无需实验室的即时检测,Piepenburg等还设计了用于侧向流试纸条检测的探针。该探针与荧光探针相似,其正向引物标记羧基荧光素(Carboxyfluorescein,FAM),反向引物标记生物素,因此,在Nfo和Bsu聚合酶的作用下,最终生成带有双标记的扩增产物。在试纸条上,扩增产物先与带有FAM抗体的胶体金结合,再在检测线上被生物素抗体捕捉,使检测线显色,达到检测目的。目前,英国公司TwistDX Inc基于以上两种探针,已分别开发出TwistAmp® exo试剂盒和TwistAmp® nfo试剂盒。TwistAmp® exo试剂盒是用于荧光检测,不过将Nfo替换成了核酸外切酶Ⅲ(exo),而TwistAmp® nfo试剂盒是用于试纸条检测。目前,RPA已经可以替代传统PCR实现检测,特别是在病毒检测方面颇具竞争力,如裂谷热病毒[48],检出限达到19个拷贝;艰难梭状芽孢杆菌[49],检出限为1 000个DNA拷贝,相当于1fg DNA;布鲁氏菌[50],检出限可低至3个拷贝。
RPA其优点在于:反应全程等温进行,不需热变性;引物设计简单;灵敏度高,最低能检测出仅含一个拷贝的样品;特异性强,可进行多重RPA。RPA的主要缺点:由于引物(30-35 nt)与探针(46-54 nt)较长,不适合较短靶序列的检测[5];RPA反应条件较苛刻,不适合直接扩增粗样品[7]。
1.6 信号介导RNA扩增技术Wharam等[51]于2001年创建了信号介导RNA扩增技术(Signal-mediated amplification of RNA technology,SMART)。SMART与NASBA一样,是一种依赖RNA转录的等温扩增技术。但是,SMART不是通过增加靶序列拷贝数实现检测,而是通过信号的放大来进行。
SMART体系含有两条探针,分别是延伸探针和模板探针。两条探针均有一个区域可分别与靶序列互补结合,同时还含有另一个可相互互补结合的区域,从而两条探针与靶序列形成SMART反应中关键的三通结构(Three-way junction,3WJ)。延伸探针在Bst DNA聚合酶的作用下,以模板探针为模板,形成双链T7 RNA聚合酶启动子和双链转录模板。双链启动子激活T7 RNA聚合酶不断对转录模板进行转录,线性合成RNA信号。为了获得更佳的检测灵敏度,可进一步扩增RNA信号,将已生成的RNA信号与另一条探针结合,该探针结构与模板探针结构相似,也有T7 RNA聚合酶启动子以及转录模板。后续的扩增过程与先前的扩增相同,双链T7 RNA聚合酶启动子和双链转录模板生成,T7 RNA聚合酶不断对转录模板进行转录,线性合成新的RNA信号。RNA信号用酶联寡核苷酸吸附实验(Enzyme-linked oligosorbent assay,ELOSA)进行检测[51]。
SMART是一种特殊的等温扩增技术,通过扩增信号来检测靶序列。Wharam等使用SMART成功检测大肠杆菌基因组DNA和总RNA中的特异性序列,甚至不需要提取核酸,直接进行检测。Hall等[52]选用g20基因,使用SMART成功区分海洋噬藻体的两个种——S-PM2和S-BnM1,证明SMART可直接检测粗样品。Murakami等[53]首次将3WJ结构与PG-RCA联合使用,成功检测人CD4mRNA,其中PG-RCA起着信号放大的作用;周敏等[54]使用相同的方法也成功检测到肠道病毒71型,其检测限低至埃摩尔级,灵敏度极强。经实验验证,SMART的主要优点为:检测范围广,既可检测DNA,也可检测RNA;探针设计简单,只需设计与靶序列结合的区域,其它的都是通用序列区域;特异性强,只有形成3WJ结构,RNA信号才能生成;可直接检测粗提样品[55]。由于SMART还存在较多的不足,如反应时间长、靶序列须是单链核酸片段、操作繁杂、灵敏度较低等,目前相关的应用报道还较少。SMART在接下里的发展中,应该专注于提高检测灵敏度,发展比ELOSA更高效的检测方法,以及将扩增与检测集中于一个试管中[5, 51]。
1.7 单引物等温扩增单引物等温扩增技术(Single primer isothermal amplification,SPIA)由Kurn等[56]于2005年首次报道。该技术主要通过使用一条嵌合引物“5'-RNA-DNA-3'”、RNase H和具有强链置换活性的DNA聚合酶来实现核酸等温扩增。
反应初期,嵌合引物与单链DNA模板互补结合,在DNA聚合酶的聚合作用下开始延伸。同时,RNase H降解嵌合引物与模板形成的杂合链DNA:RNA中的RNA链[57],使未结合引物RNA部分与模板结合,在DNA聚合酶的链置换活性下,新引物置换旧引物的延伸产物,并开始延伸。与此同时,新引物与模板形成的杂合链中的RNA链再次被RNase H降解,如此循环,进行线性扩增,从而产生大量与单链DNA模板互补的单链DNA产物。若想限制产物的长度,可使用链终止多聚核苷酸(Blocker)。Blocker能与单链DNA模板结合,当引物延伸至blocker时,将停止延伸,这是因为blocker中的部分碱基已被修饰,其与模板的结合力大大增强,DNA聚合酶无法置换出blocker。为防止blocker发生延伸反应,其3'端也需要进行修饰[18, 56]。在SPIA的基础上,通过添加逆转录酶,可实现以RNA为模板扩增DNA,该技术被称为Ribo-SPIA,其检出限为1 ng RNA,且具有很好的重复性[56]。
SPIA的主要优点在于:使用单引物,减少了引物二聚体的形成;不受RNA干扰;反应机制简单,效率高。目前,其已被用于基于表达分析以及病原菌检测[58-60],同时证明该技术在检测沙门氏菌方面比qPCR(quantitative PCR,qPCR)具有更好的灵敏度和特异性。但是,SPIA也存在其缺点:模板是单链核酸,需对双链核酸进行热变性处理;需对blocker进行碱基修饰,提高扩增成本;引物是嵌合引物,设计较复杂。
1.8 嵌合引物引发的核酸等温扩增嵌合引物引发的核酸等温扩增技术(Isothermal and chimeric primer-initiated amplification of nucleic acids,ICAN)[61, 62]与SPIA相似,均主要依靠嵌合引物、RNase H和具有强链置换活性的DNA聚合酶,不过ICAN需要一对引物,同时引物中的DNA与RNA序列排布位置与SPIA的引物相反,是“5'-DNA-RNA-3'”构型。ICAN的扩增产量高度依赖引物浓度,随着浓度的增高,产量显著提升。
重复“nick-and-run”是Mukai等[62]对ICAN扩增机制提出的第一个假设。一条引物与模板结合后,在DNA聚合酶的作用下开始延伸。随即,RNase H切割引物RNA部分与引物延伸产物之间的结合点,形成缺口。DNA聚合酶再次作用于RNA 3'端,在置换出下游DNA的同时,引物开始重新延伸,结合点再次形成。RNase H再次切割结合点,在DNA聚合酶的作用下,引物继续重新延伸,进入循环扩增。得到的单链DNA则作为另一条引物的模板进行扩增,原理相同。依照重复“nick-and-run”扩增机制,扩增产物中不应含有RNA残基,然而经序列分析,发现扩增产物中存在着大量带有RNA残基的产物。因此,Uemori等[63]认为ICAN还存在着其它反应机制,他们分别提出了“multi-priming”和“template-switching”两种新机制。经研究发现,RNase H引物存在2个或更多RNA残基时,RNase H会优先切割RNA残基5'端,并且是从引物上最后一个RNA残基开始一个一个切割,即使只剩最后1个RNA残基,RNase H也能有效切割残基的5'端。但是,“multi-priming”和“template-switching”两种新机制都是基于只切割引物上最后一个RNA残基5'端建立起来的,因此,实际上,ICAN的扩增机制可能会更加复杂。
与其它核酸扩增技术一样,ICAN也可用于病原菌检测。Isogai等[64]使用ICAN通过扩增invA基因来检测死鸡、蛋黄和牛粪中的沙门氏菌,且在死鸡冲洗液的检测中,ICAN的检出率高于PCR。Horii等[65]结合层析技术,应用ICAN成功快速检测出淋球菌对氟喹诺酮类药物的抗药性。Urasaki等[66]在ICAN的基础上,引入环状探针,对44个样品中的柑橘黄龙病菌进行检测,相比常规PCR,ICAN的检测时间可以至少节约2 h,同时灵敏度更高。
综上,ICAN的主要优点在于扩增效率高、稳定性好、扩增对象范围广,除了双链DNA,还可作用于cDNA:RNA杂合链。但是,由于ICAN使用的引物是嵌合引物,由RNA和DNA构成,合成较复杂,且扩增机制不明确,进而不利于该技术的发展。
2 依赖于切刻内切酶的等温扩增技术解决不依赖高温使DNA模板变性进而引发扩增反应,是实现真正等温扩增反应的瓶颈。虽然,DNA解旋酶等被用来产生DNA单链,但是效果一般,反应机制复杂。切刻内切酶的发现,使得等温产生DNA单链成为可能。切刻内切酶(Nicking Enzyme),也称为切口酶,由美国NEB公司开发,其为一种限制性内切酶,能够识别特异性的核苷酸序列,并且在序列内部或首尾处发生单链切割;该过程对底物DNA没有要求,即不需要有任何化学修饰。因此,依赖切刻内切酶作为反应第一步的等温扩增技术逐渐被发展起来。
2.1 链置换扩增技术(Strand displacement ampli-fication,SDA)置换扩增技术是一种DNA体外等温扩技术,由美国Beetonniekinson研究中心Walke[67]博士团队于1992年首次报道,此方法的建立标志着一种新型DNA扩增技术的诞生。
SDA反应全过程主要依赖于限制性内切酶对半硫代磷酸化碱基对应互补链的切割作用,以及聚合酶exo-klenow对切口的延伸作用和对下游DNA片段的置换作用。SDA由两条引物P1和P2进行扩增,两条引物的3'端和5'端分别含有特异性结合序列和Hinc Ⅱ识别序列,其具体的扩增方式是:限制性内切酶首先识别半硫代磷酸化碱基对应的互补链,然后切割该位置并产生单链切口;被切割部分的5'上游端能够作为一条引物,在具有链置换活性的DNA聚合酶的作用下从5'端向3'端延伸扩增,从而剥离原本已经形成互补的DNA链;被取代下的单链DNA再与另一条引物结合,相同原理下延伸形成一条新的双链DNA。SDA反应即是通过这种“切口-扩增-置换”循环往复的过程,达到靶序列高效扩增的目的,其一般反应条件为37-40℃,经过2 h循环靶序列可得到108倍的扩增量。
SDA被发明后,在20余年的发展更新中,该技术在细菌病毒检测、核酸定量、重金属检测及芯片杂交等多个方面有相关报道,同时也衍生出了一些新方法。Little等[68]将SDA与荧光共振能量转移技术结合,开发出新一代DNA探针系统—BD ProbeTecTM ET系统,用于检测沙眼衣原体、淋病奈瑟菌和人乳头状瘤病毒;Nuovo等[69]应用RT-SDA检测丙型肝炎病毒;Li等[70]研发出一种SDA结合核酸酶高效检测二价铅离子的方法。
SDA提供了一种可用于核酸诊断分析的扩增方法,具有等温核酸扩增的通用优势:无须热变性、特异高效、操作简便,只需一个恒温器,大大简化了对所需仪器的要求。但是SDA由于反应原理和反应体系的复杂性,不可避免的带来了很多缺点:非标准核苷酸的使用,经硫基修饰的单核苷酸在价格上比普通的单核苷酸高出很多倍,增加了反应成本[71];经硫基修饰的单核苷酸不是DNA聚合酶的天然底物,修饰的核苷酸与标准核苷酸存在竞争关系,聚合酶容易从DNA模板上掉落,大大降低了扩增效率降低了扩增效率,所以,SDA不可能合成较长的产物,一般不超过200 bp[18, 72];SDA产物末端带有限制性核酸内切酶的识别序列或其残端,使其不适合直接用于克隆,因此,该技术在基因工程方面不占优势[73];SDA的等温过程是两步法,需要首先有变温过程打开双链与引物退火,这就需要酶体系在变温环节后添加,增加了外源污染的可能性。
2.2 指数扩增反应(Exponential amplification reaction,EXPAR)指数扩增反应由Van Ness等[74]于2003年开发,该技术利用扩增模板合成寡核苷酸,长度为8-16 mer,可用于位点多态性研究[7]。反应试剂中的N.BstNBI切口酶在反应中发挥着至关重要的作用,因此,也可以把该技术称为切口酶扩增反应(Nicking enzyme amplification,NEAR)[7]。
EXPAR能否顺利开展,取决于触发反应。而触发反应存在着不同的机制,其中最简单的一种机制是依赖基因组DNA上存在着切口酶的识别序列。单链基因组DNA与触发模板退火结合,双链识别序列形成,激活切口酶切割单链基因组DNA识别序列,缺口下游带有3'悬挂的DNA序列,其在60℃下自动解离,同时上游DNA序列以触发模板为模板,在DNA聚合酶的作用下开始延伸,合成平末端的DNA序列。与此同时,新形成的识别序列再次被切割,缺口下游的DNA序列自动解离,成为触发子。上游DNA序列继续延伸,切口酶继续切割,从而线性生成触发子,反应进入指数扩增阶段。形成的触发子与扩增模板3'端序列结合,在DNA聚合酶的作用下开始延伸,形成双链DNA,切口酶切割识别序列,形成缺口,缺口下游序列自动解离,解离的单链片段随即与自由的扩增模板结合形成短暂双链结构,在DNA聚合酶的作用下,解离的片段一旦开始延伸,双链结构就会稳定下来,不会轻易解离,最终生成新的双链DNA。新生成的双链DNA重复之前的过程,继续合成其它新的双链DNA。在这个过程中,每个双链DNA也会线性生成单链片段,从而达到指数扩增的目的。
EXPAR发展至今,已被广泛应用于检测蛋白质和DNA。Nie等[75]基于EXPAR,开发出GQ-EXPAR,这是一种依赖G-四链体的双重扩增技术,可通过联合比色法检测DNA。Ma等[76]创建了一种用于检测转录因子的技术,该技术是在GQ-EXPAR的基础上,再加入RNA转录技术实现检测的目的。Yu等[77]联合EXPAR和HCR开发出一种电化学传感器,可对H7N9病毒实现超灵敏检测。EXPAR的主要优点在于:反应形式多,可满足不同的扩增需求;反应稳定性好,速度快,灵敏度高。但由于反应机制复杂,产物产量易受扩增模板限制,使得后期会从指数扩增转变为线性扩增。
2.3 切刻内切酶介导等温核酸扩增技术(Nicking enzyme mediated amplification,NEMA)切刻内切酶介导等温核酸扩增技术是2006年研发的一种全新的核酸恒温扩增技术,该技术是在SDA的基础上发展而来的不依赖于特殊化学修饰的等温方法[72]。NEMA反应体系中使用的切刻内切酶,是一种能够识别特异性的位点并发生自然单链切割产生缺口的酶,切刻内切酶的使用克服了传统SDA反应中需要添加化学修饰的非标准核苷酸的缺陷,简化了反应体系,提高了反应效率,成本更低,并且可合成长链DNA。
NEMA反应体系包括两对引物(剥离引物B1/B2,切割引物S1/S2)、一种具有链置换活性的DNA聚合酶、一种能够识别特异性切割位点的切刻内切酶、额外添加的镁离子以及适宜的缓冲溶液。NEMA技术是建立在SDA的原理技术上改进发明的,因此二者基本扩增原理相似。NEMA扩增分为“切割单链形成切口”和“链置换剥离旧链”两个过程,在待扩增的DNA双链模板上有一段切口酶特异识别的序列,在切口酶的作用下只切割其中一条链,然后DNA聚合酶以形成的切口为起点,以未被切断的单链为引物延伸形成新链,置换旧链,产物作为新循环的模板不断参与新一轮的指数扩增。体系中还存在另外一种模板,就是只上游或者下游一端引入了切刻内切酶识别位点的序列,这种状态的靶序列同样是以不断结合杂交互补的切割引物参与扩增,然后发生单链切口切割,在DNA聚合酶作用下链置换,如此的往复循环过程。
相较于SDA和其他目前开发的等温核酸技术,NEMA具有一些潜在的优势:酶体系和原材料体系的简单减少了抑制因子的干扰;扩增迅速,反应时间短,在1 h之内就能达到电泳凝胶的观察要求;低成本,操作简单,对操作仪器的要求不高,为核酸扩增技术的推广创造了条件。但是NEMA目前还处于探索阶段,多数应用在微生物质粒DNA的检测方面。
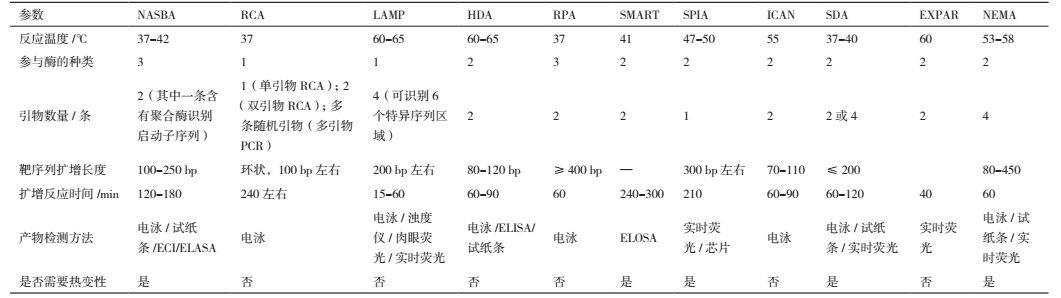
3 结语本文介绍了10余种等温扩增技术,其中每种技术均有各自的优势,表 1比较了上文提到的几种等温核酸扩增方法的技术特点。尽管等温扩增技术有着普通PCR扩增不可超越的优势,但是有些缺点也是不容忽视的。不过,随着进一步研究,对生物过程的了解越来越深入,相信未来将会有更多更完善的新型等温扩增技术不断涌现。
等温扩增技术是在单一温度下对靶序列进行扩增或信号放大,不依赖便携性差、价格昂贵的热循环仪,进而在近20多年得到快速发展,尤其是在微生物检测领域中。我国现行标准(GB/T,SN/T等)中有多项采用了等温检测的方法,包括金黄色葡萄球菌、产气荚膜梭菌、创伤弧菌、阪崎肠杆菌、沙门氏菌等致病菌;猪圆环病毒2型、家畜布鲁氏菌、焦枯病菌、甘蔗病原菌和猪瘟病毒等。等温扩增技术对仪器要求简单,仅以电池为电源的加热器即可,未来可在边远地区和基层医疗单位等缺乏资金和先进检测设备的地区大力推广,使其具备快速诊断病原体的能力。检测时间短也是等温扩增技术的优势,可在实时现场检测与监测领域推广,如在突发公共卫生事件的现场诊断中,等温技术对传染性疾病的早期发现和控制具有重要的意义。
由于等温扩增技术的等温反应的特点,其很有可能普遍应用于下一代现场快速检测设备中。然而,历经了20余年的发展,在市场上仍然看不到任何已经被广泛使用的相关设备。但是,导致这种现象的原因并非等温扩增技术不适用,而是因为这是一个复杂的工程问题,是一个涉及多领域的难题[78]。想研发出以核酸为靶物质的便携快速即时检测设备,我们需要对等温扩增技术进行更多的优化,比如扩增速率,电力需求等等,同时需要解决将等温扩增与上下游操作结合在一起的技术难关,如结合样品核酸的提取纯化以及后期的扩增产物检测[79]。随着微型品制造技术与等温扩增技术的不断提升[7],相信在不久的将来将会出现突破性的进展。
| [1] | Zhao Y, Chen F, Li Q, et al. Isothermal amplification of nucleic acids[J]. Chemical Reviews, 2015, 115(22): 12491–12545. DOI:10.1021/acs.chemrev.5b00428 |
| [2] | Compton J. Nucleic acid sequence-based amplification[J]. Nature, 1991, 350(6313): 91–92. DOI:10.1038/350091a0 |
| [3] | Kwoh DY, Davis GR, Whitfield KM, et al. Transcription-based amplification system and detection of amplified human immunodeficiency virus type 1 with a bead-based sandwich hybridization format[J]. Proceedings of the National Academy of Sciences of the United States of America, 1989, 86(4): 1173–1177. DOI:10.1073/pnas.86.4.1173 |
| [4] | Guatelli JC, Whitfield KM, Kwoh DY, et al. Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication[J]. Proceedings of the National Academy of Sciences, 1990, 87(5): 1874–1878. DOI:10.1073/pnas.87.5.1874 |
| [5] | Li J, Macdonald J. Advances in isothermal amplification:novel strategies inspired by biological processes[J]. Biosensors & Bioelectronics, 2015, 64: 196–211. |
| [6] | Deiman B, van Aarle P, Sillekens P. Characteristics and applications of nucleic acid sequence-based amplification(NASBA)[J]. Molecular Biotechnology, 2002, 20(2): 163–179. DOI:10.1385/MB:20:2 |
| [7] | Deng H, Gao Z. Bioanalytical applications of isothermal nucleic acid amplification techniques[J]. Analytica Chimica Acta, 2015, 853: 30–45. DOI:10.1016/j.aca.2014.09.037 |
| [8] | Gill P, Ghaemi A. Nucleic Acid Isothermal Amplification Technologies—a review[J]. Nucleosides, Nucleotides and Nucleic Acids, 2008, 27(3): 224–243. DOI:10.1080/15257770701845204 |
| [9] | 汪琳, 罗英, 周琦, 等. 核酸恒温扩增技术研究进展[J]. 生物技术通讯, 2011, 22(2): 296–302. |
| [10] | Lau LT, Reid SM, King DP, et al. Detection of foot-and-mouth disease virus by nucleic acid sequence-based amplification(NASBA)[J]. Veterinary Microbiology, 2008, 126(1): 101–110. |
| [11] | Zhao X, Dong T, Yang Z, et al. Compatible immuno-NASBA LOC device for quantitative detection of waterborne pathogens:design and validation[J]. Lab on a Chip, 2012, 12(3): 602–612. DOI:10.1039/C1LC20836E |
| [12] | Clancy E, Coughlan H, Higgins O, et al. Development of internally controlled duplex real-time NASBA diagnostics assays for the detection of microorganisms associated with bacterial meningitis[J]. Journal of Microbiological Methods, 2016, 127: 197–202. DOI:10.1016/j.mimet.2016.06.017 |
| [13] | H?nsvall BK, Robertson LJ. Real-time nucleic acid sequence-based amplification(NASBA)assay targeting MIC1 for detection of Cryptosporidium parvum and Cryptosporidium hominis oocysts[J]. Experimental Parasitology, 2017, 172: 61–67. DOI:10.1016/j.exppara.2016.12.009 |
| [14] | Zeng W, Yao W, Wang Y, et al. Molecular detection of genotype Ⅱ grass carp reovirus based on nucleic acid sequence-based amplification combined with enzyme-linked immunosorbent assay(NASBA-ELISA)[J]. Journal of Virological Methods, 2017, 243: 92–97. DOI:10.1016/j.jviromet.2017.02.001 |
| [15] | Fire A, Xu SQ. Rolling replication of short DNA circles[J]. Proceedings of the National Academy of Sciences, 1995, 92(10): 4641–4645. DOI:10.1073/pnas.92.10.4641 |
| [16] | Liu D, Daubendiek SL, Zillman MA, et al. Rolling circle DNA synthesis:small circular oligonucleotides as efficient templates for DNA polymerases[J]. Journal of the American Chemical Society, 1996, 118(7): 1587–1594. DOI:10.1021/ja952786k |
| [17] | Blanco L, Bernad A, Lázaro JM, et al. Highly efficient DNA synthesis by the phage Φ29 DNA polymerase. Symmetrical mode of DNA replication[J]. Journal of Biological Chemistry, 1989, 264(15): 8935–8940. |
| [18] | 彭涛. 核酸等温扩增技术及其应用[D]. 北京: 科学出版社, 2009. |
| [19] | Lizardi PM, Huang X, Zhu Z, et al. Mutation detection and single-molecule counting using isothermal rolling-circle amplification[J]. Nature Genetics, 1998, 19(3): 225–232. DOI:10.1038/898 |
| [20] | Dean FB, Nelson JR, Giesler TL, et al. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification[J]. Genome Research, 2001, 11(6): 1095–1099. DOI:10.1101/gr.180501 |
| [21] | 何艳, 蒋涛. 基于链置换反应的DNA等温扩增技术应用进展[J]. 医学综述, 2010, 16(1): 24–27. |
| [22] | Daubendiek SL, Ryan K, Kool ET. Rolling-circle RNA synthesis:circular oligonucleotides as efficient substrates for T7 RNA polymerase[J]. Journal of the American Chemical Society, 1995, 117(29): 7818–7819. DOI:10.1021/ja00134a032 |
| [23] | Dean FB, Hosono S, Fang L, et al. Comprehensive human genome amplification using multiple displacement amplification[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(8): 5261–5266. DOI:10.1073/pnas.082089499 |
| [24] | Spits C, Le Caignec C, De Rycke M, et al. Optimization and evaluation of single-cell whole-genome multiple displacement amplification[J]. Human Mutation, 2006, 27(5): 496–503. DOI:10.1002/(ISSN)1098-1004 |
| [25] | Luthra R, Medeiros LJ. Isothermal multiple displacement amplification:a highly reliable approach for generating unlimited high molecular weight genomic DNA from clinical specimens[J]. The Journal of Molecular Diagnostics, 2004, 6(3): 236–242. DOI:10.1016/S1525-1578(10)60516-8 |
| [26] | Rockett R, Barraclough KA, Isbel NM, et al. Specific rolling circle amplification of low-copy human polyomaviruses BKV, HPyV6, HPyV7, TSPyV, and STLPyV[J]. Journal of Virological Methods, 2015, 215: 17–21. |
| [27] | Wen J, Li W, Li J, et al. Study on rolling circle amplification of Ebola virus and fluorescence detection based on graphene oxide[J]. Sensors and Actuators B:Chemical, 2016, 227: 655–659. DOI:10.1016/j.snb.2016.01.036 |
| [28] | Hao L, Gu H, Duan N, et al. An enhanced chemiluminescence resonance energy transfer aptasensor based on rolling circle amplification and WS 2 nanosheet for Staphylococcus aureus detection[J]. Analytica Chimica Acta, 2017, 959: 83–90. DOI:10.1016/j.aca.2016.12.045 |
| [29] | Gusev Y, Sparkowski J, Raghunathan A, et al. Rolling circle amplification:a new approach to increase sensitivity for immunohistochemistry and flow cytometry[J]. American Journal of Pathology, 2011, 159(1): 63–69. |
| [30] | Notomi T, Okayama H, Masubuchi H, et al. Loop-mediated isothermal amplification of DNA[J]. Nucleic Acids Research, 2000, 28(12): e63. DOI:10.1093/nar/28.12.e63 |
| [31] | Nagamine K, Watanabe K, Ohtsuka K, et al. Loop-mediated isothermal amplification reaction using a nondenatured template[J]. Clinical Chemistry, 2001, 47(9): 1742–1743. |
| [32] | Kaneko H, Kawana T, Fukushima E, et al. Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances[J]. Journal of Biochemical and Biophysical Methods, 2007, 70(3): 499–501. DOI:10.1016/j.jbbm.2006.08.008 |
| [33] | Nagamine K, Hase T, Notomi T. Accelerated reaction by loop-mediated isothermal amplification using loop primers[J]. Molecular and Cellular Probes, 2002, 16(3): 223–229. DOI:10.1006/mcpr.2002.0415 |
| [34] | Aonuma H, Yoshimura A, Kobayashi T, et al. A single fluorescence-based LAMP reaction for identifying multiple parasites in mosquitoes[J]. Experimental Parasitology, 2010, 125(2): 179–183. DOI:10.1016/j.exppara.2009.12.023 |
| [35] | He L, Xu H. Development of a multiplex loop-mediated isothermal amplification(mLAMP)method for the simultaneous detection of white spot syndrome virus and infectious hypodermal and hematopoietic necrosis virus in penaeid shrimp[J]. Aquaculture, 2011, 311(1): 94–99. |
| [36] | Liang C, Chu Y, Cheng S, et al. Multiplex loop-mediated isothermal amplification detection by sequence-based barcodes coupled with nicking endonuclease-mediated pyrosequencing[J]. Analytical Chemistry, 2012, 84(8): 3758–3763. DOI:10.1021/ac3003825 |
| [37] | Tanner NA, Zhang Y, Evans Jr TC. Simultaneous multiple target detection in real-time loop-mediated isothermal amplification[J]. Biotechniques, 2012, 53(2): 81–89. |
| [38] | Chen Y, Cheng N, Xu Y, et al. Point-of-care and visual detection of P. aeruginosa and its toxin genes by multiple LAMP and lateral flow nucleic acid biosensor[J]. Biosensors and Bioelectronics, 2016, 81: 317–323. DOI:10.1016/j.bios.2016.03.006 |
| [39] | Vincent M, Xu Y, Kong H. Helicase-dependent isothermal DNA amplification[J]. EMBO Reports, 2004, 5(8): 795–800. DOI:10.1038/sj.embor.7400200 |
| [40] | An L, Wen T, Ranalli TA, et al. Characterization of a thermostable UvrD helicase and its participation in helicase dependent amplifi-cation[J]. Journal of Biological Chemistry, 2005, 280(32): 28952–28958. DOI:10.1074/jbc.M503096200 |
| [41] | Li Y, Kumar N, Gopalakrishnan A, et al. Detection and species identification of malaria parasites by isothermal tHDA amplification directly from human blood without sample preparation[J]. Journal of Molecular Diagnostics, 2013, 15(5): 634–641. DOI:10.1016/j.jmoldx.2013.05.005 |
| [42] | Motré A, Li Y, Kong H. Enhancing helicase-dependent amplification by fusing the helicase with the DNA polymerase[J]. Gene, 2008, 420(1): 17–22. DOI:10.1016/j.gene.2008.04.017 |
| [43] | Xu Y, Kim HJ, Kays A, et al. Simultaneous amplification and scr-eening of whole plasmids using the T7 bacteriophage replisome[J]. Nucleic Acids Research, 2006, 34(13): e98–e98. DOI:10.1093/nar/gkl547 |
| [44] | Goldmeyer J, Kong H, Tang W. Development of a Novel one-tube isothermal reverse transcription thermophilic helicase-dependent amplification platform for rapid RNA detection[J]. The Journal of Molecular Diagnostics, 2007, 9(5): 639–644. DOI:10.2353/jmoldx.2007.070012 |
| [45] | Piepenburg O, Williams CH, Stemple DL, et al. DNA detection using recombination proteins[J]. PLoS Biology, 2006, 4(7): e204. DOI:10.1371/journal.pbio.0040204 |
| [46] | 吕蓓, 程海荣, 严庆丰, 等. 用重组酶介导扩增技术快速扩增核酸[J]. 中国科学:生命科学, 2010, 40(10): 983–988. |
| [47] | Liu J, Morrical SW. Assembly and dynamics of the bacteriophage T4 homologous recombination machinery[J]. Virology Journal, 2010, 7(1): 128–133. DOI:10.1186/1743-422X-7-128 |
| [48] | Euler M, Wang Y, Nentwich O, et al. Recombinase polymerase amplification assay for rapid detection of Rift Valley fever virus[J]. Journal of Clinical Virology, 2012, 54(4): 308–312. DOI:10.1016/j.jcv.2012.05.006 |
| [49] | Tsaloglou MN, Watson RJ, Rushworth CM, et al. Real-time microfluidic recombinase polymerase amplification for the toxin B gene of Clostridium difficile on a SlipChip platform[J]. Analyst, 2015, 140(1): 258–264. DOI:10.1039/C4AN01683A |
| [50] | Hang R, Yang M, Zhang G, et al. Development of a rapid recombinase polymerase amplification assay for detection of Brucella in blood samples[J]. Molecular & Cellular Probes, 2016, 30(2): 122–124. |
| [51] | Wharam SD, Marsh P, Lloyd JS, et al. Specific detection of DNA and RNA targets using a novel isothermal nucleic acid amplification assay based on the formation of a three-way junction structure[J]. Nucleic Acids Research, 2001, 29(11): e54. DOI:10.1093/nar/29.11.e54 |
| [52] | Hall MJ, Wharam SD, Weston A, et al. Use of signal-mediated amplification of RNA technology(SMART)to detect marine cyanophage DNA[J]. Biotechniques, 2002, 32(3): 604–611. |
| [53] | Murakami T, Sumaoka J, Komiyama M. Sensitive RNA detection by combining three-way junction formation and primer generation-rolling circle amplification[J]. Nucleic Acids Research, 2012, 40(3): e22. DOI:10.1093/nar/gkr909 |
| [54] | 周敏, 张宏萍, 陆仁飞, 等. 三向连接构造组合引物介导的滚环扩增技术检测肠道病毒71型方法的建立[J]. 山东医药, 2015(28): 21–23. DOI:10.3969/j.issn.1002-266X.2015.28.007 |
| [55] | Yan L, Zhou J, Zheng Y, et al. Isothermal amplified detection of DNA and RNA[J]. Molecular Biosystems, 2014, 10(5): 970–1003. DOI:10.1039/c3mb70304e |
| [56] | Kurn N, Chen P, Heath JD, et al. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications[J]. Clinical Chemistry, 2005, 51(10): 1973–1981. DOI:10.1373/clinchem.2005.053694 |
| [57] | 何水林. 基因工程[M]. 北京: 科学出版社, 2008. |
| [58] | Barker CS, Griffin C, Dolganov GM, et al. Increased DNA microarray hybridization specificity using sscDNA targets[J]. BMC Genomics, 2005, 6(1): 1–8. DOI:10.1186/1471-2164-6-1 |
| [59] | Guo X, Guo Y, Yan S, et al. A new molecular diagnosis method combined single primer isothermal amplification with rapid isothermal detection assay in detection of group B Streptococcus[J]. African Journal of Microbiology Research, 2013, 7(34): 4317–4322. |
| [60] | Wang J, Rui L, Hu L, et al. Development of a quantitative fluorescence single primer isothermal amplification-based method for the detection of Salmonella[J]. International Journal of Food Microbiology, 2015, 219: 22–27. |
| [61] | Mukai H, Sagawa H, Uemori T, et al. Method for amplifying nucleic acid sequence:US, 6951722[P]. 2005-10-4. |
| [62] | Mukai H, Uemori T O, Kobayashi E, et al. Highly efficient isothermal DNA amplification system using three elements of 5'-DNA-RNA-3' chimeric primers, RNaseH and strand-displacing DNA polymerase[J]. Journal of Biochemistry, 2007, 142(2): 273–281. DOI:10.1093/jb/mvm138 |
| [63] | Uemori T, Mukai H O, Moriyama M, et al. Investigation of the molecular mechanism of ICAN, a novel gene amplification method[J]. Journal of Biochemistry, 2007, 142(2): 283–292. DOI:10.1093/jb/mvm137 |
| [64] | Isogai E, Makungu C, Yabe J, et al. Detection of Salmonella invA by isothermal and chimeric primer-initiated amplification of nucleic acids(ICAN)in Zambia[J]. Comparative Immunology Microbiology & Infectious Diseases, 2005, 28(5): 363–370. |
| [65] | Horii T, Monji A, Uemura K, et al. Rapid detection of fluoroquino-lone resistance by isothermal chimeric primer-initiated amplifica-tion of nucleic acids from clinical isolates of Neisseria gonorrho-eae[J]. Journal of Microbiological Methods, 2006, 65(3): 557–561. DOI:10.1016/j.mimet.2005.10.002 |
| [66] | Urasaki N, Kawano S, Mukai H, et al. Rapid and sensitive detection of "Candidatus Liberibacter asiaticus" by cycleave isothermal and chimeric primer-initiated amplification of nucleic acids[J]. Journal of General Plant Pathology, 2008, 74(2): 151–155. DOI:10.1007/s10327-008-0083-7 |
| [67] | Walker GT, Little MC, Nadeau JG, et al. Isothermal in vitro amplification of DNA by a restriction enzyme/DNA polymerase system[J]. Proceedings of the National Academy of Sciences of the United States of America, 1992, 89(1): 392–396. DOI:10.1073/pnas.89.1.392 |
| [68] | Little MC, Andrews J, Moore R, et al. Strand displacement amplification and homogeneous real-time detection incorporated in a second-generation DNA probe system, BDProbeTecET[J]. Clinical Chemistry, 1999, 45(6): 777–784. |
| [69] | Nuovo GJ. In situ strand displacement amplification:an improved technique for the detection of low copy nucleic acids[J]. Diagnostic Molecular Pathology, 2000, 9(4): 195–202. DOI:10.1097/00019606-200012000-00004 |
| [70] | Li W, Yang Y, Chen J, et al. Detection of lead(Ⅱ)ions with a DNAzyme and isothermal strand displacement signal amplification[J]. Biosensors and Bioelectronics, 2014, 53: 245–249. DOI:10.1016/j.bios.2013.09.055 |
| [71] | Yao Z, Lidgard G. Methods for rapid, single-step strand displacem-ent amplification of nucleic acids:US, 11/838, 024[P]. 2007-8-13. |
| [72] | 尤其敏, 净汪, 林胡, 等. 切口酶扩增靶核酸序列的方法及用于扩增靶核酸序列的试剂盒及其应用: 中国, 200610057262[P]. 2006-10-25. |
| [73] | 马丽敏, 卢亦愚. 核酸等温扩增技术研究进展[J]. 浙江预防医学, 2013, 25(1): 24–27. |
| [74] | Van Ness J, Van Ness LK, et al. Isothermal reactions for the amplification of oligonucleotides[J]. Proceedings of the National Academy of Sciences, 2003, 100(8): 4504–4509. DOI:10.1073/pnas.0730811100 |
| [75] | Nie J, Zhang DW, Cai T, et al. G-quadruplex based two-stage isothermal exponential amplification reaction for label-free DNA colorimetric detection[J]. Biosensors & Bioelectronics, 2014, 56: 237–242. |
| [76] | Ma F, Yang Y, Zhang CY. Ultrasensitive detection of transcription factors using transcription-mediated isothermally exponential amplification-induced chemiluminescence[J]. Analytical Chemistry, 2014, 86(12): 6006–6011. DOI:10.1021/ac5017369 |
| [77] | Yu Y, Chen Z, Jian W, et al. Ultrasensitive electrochemical detection of avian influenza A(H7N9) virus DNA based on isothermal exponential amplification coupled with hybridization chain reaction of DNAzyme nanowires[J]. Biosensors and Bioelectronics, 2015, 64: 566–571. DOI:10.1016/j.bios.2014.09.080 |
| [78] | Craw P, Balachandran W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics:a critical review[J]. Lab on a Chip, 2012, 12(14): 2469–2486. DOI:10.1039/c2lc40100b |
| [79] | Singh R, Maganti RJ, Jabba SV, et al. Microarray-based comparison of three amplification methods for nanogram amounts of total RNA[J]. American Journal of Physiology-Cell Physiology, 2005, 288(5): C1179–C1189. DOI:10.1152/ajpcell.00258.2004 |