



含有环己基的手性醇是很多医药、农药、有机化工产品及材料的重要中间体[1],譬如,合成酸增殖剂的关键前体1-苯基-cis-1,2-环己二醇,合成手性药物的手性砌块光学纯的β-氨基环己醇等。利用基因工程技术构建不对称还原取代环己酮的生物催化剂,为合成这类手性醇提供了一种安全性、环境相容性、立体选择性均高的方法,但是,目前关于这方面的报道较少[2]。糖多孢红霉菌聚酮合成酶的6个模块中有5个酮还原酶(EryKR),是一个催化羰基不对称还原的天然酶库。酮还原酶表现出广泛的底物谱:酮还原酶不仅能还原天然底物链式聚酮链及与其相似的底物如(2R,S)-2-甲基-3-羰基戊酰N-乙酰半胱酰胺硫酸酯等[3, 4],而且还能以完全不同于链式聚酮链的含环己基结构的化合物作为底物[5, 6],如环己酮、2-甲基环己酮和2-烯丙基环己酮等,显示了EryKR有望成为立体选择性还原取代环己酮的手性生物催化剂的巨大潜力。
Ⅰ型聚酮合成酶的酮还原酶根据对链式聚酮链的立体选择性还原情况分为A型和B型,糖多孢红霉菌聚酮合成酶模块1的酮还原酶(EryKR1) 为B型酮还原酶,在第88-103个氨基酸处有一个LDD模式序列,而糖多孢红霉菌聚酮合成酶模块2的酮还原酶(EryKR2) 为A型酮还原酶,其中没有这个模式[7]。多种聚酮合成酶的酮还原酶对包括(2R,S)-2-甲基-3-羰基戊酰N-乙酰半胱酰胺硫酸酯在内的若干α-替代的β-羰基双酮化合物进行的立体选择性还原研究,发现若这些位点发生了突变,酮还原酶对这些底物的立体选择性会发生相应的改变,证实了这些酶中存在有控制立体选择性还原的模式序列[7, 8]。
考虑到除了链式聚酮链,酮还原酶还能以含环己基结构的化合物为底物。为了探讨LDD模式序列是否也是控制含环己基结构的化合物立体选择性还原的位点,本研究利用重叠PCR技术将EryKR1(B型KR)中控制立体选择性还原的LDD模式序列突变成EryKR2(A型KR)中的相应位点PQQ,并且将EryKR2中的PQQ突变成EryKR1中的LDD,通过比较突变型酮还原酶与野生型酮还原酶以2-甲基环己酮为底物的酶活以及还原产物的立体结构,分析2-甲基环己酮的主要还原产物的立体结构转变与LDD模式序列的变化之间的关联,确定LDD模式序列是否为酶中负责控制2-甲基环己酮立体选择性还原的位点,旨在为指导催化不对称还原取代环己酮的手性生物催化剂的构建及开发奠定理论基础。
1 材料与方法 1.1 材料 1.1.1 菌种、质粒和培养基糖多孢红霉菌(Saccharopolyspora erythraea)、大肠杆菌表达载体pET28a质粒及重组菌E.coli BL21(pET28a-gdh)为本实验室保藏;大肠杆菌BL21(E.coli BL21(DE3))为本实验宿主菌,购买于康为世纪生物科技有限公司。LB培养基[9]:酵母膏5 g/L、蛋白胨10 g/L、NaCl 10 g/L,pH7.0,用于培养大肠杆菌BL21,筛选抗性菌株时,加卡拉霉素终浓度为100 μg/mL;TSB培养基:Tryptic Soy Broth 30 g/L,用于糖多孢红霉菌的液体培养。
1.1.2 酶和化学试剂Lysozyme、蛋白酶K、pfu DNA聚合酶、限制性内切酶NdeⅠ和EcoRⅠ、T4 DNA连接酶、DNA ladder Marker、Protein Marker、DNA Loading Buffer、微量琼脂糖凝胶DNA回收试剂盒及高纯度质粒小提试剂盒均为康为世纪生物科技有限公司产品,2-甲基环己酮(98%)和反式-2-甲基环己醇(97%)为Alfa Aesar公司的产品,顺式-2-甲基环己醇(98%)购自Aldrich。
1.2 方法 1.2.1 野生型及突变型eryKR1或eryKR2基因的克隆及表达质粒的构建糖多孢红霉菌A226基因组DNA的提取参照文献[6]进行。根据文献[5]报道的EryKR1和EryKR2的边界,设计扩增野生型eryKR1基因和eryKR2基因(或突变型eryKR1即TeryKR1基因和突变型eryKR2即TeryKR2基因)的两端引物,为便于克隆,在正向引物和反向引物前分别加上NdeⅠ和EcoRⅠ酶切位点,其中用于扩增野生型和突变型eryKR1基因的两端引物为LKR1:5'-gccatatggacgaggtttccgcgctgcg-3'(gc为保护性碱基,catatg为NdeⅠ酶切位点);RKR1:5'-ggaattctcacgcgcccacccgcggttcggc-3'(g为保护性碱基,gaattc为EcoRⅠ酶切位点)。
为了采用重叠PCR技术扩增TeryKR1基因,设计中间的将EryKR1酶中的LDD对应的碱基突变成EryKR2酶中的PQQ相应的碱基的正反向引物分别TKR1-L:5'-cacgccgccgccacgccgcagcagggcaccgtggacaccctcacc-3',TKR1-R:5'-ggtgtccacggtgccctgctg cggcgtggcggcggcgtggaacac-3';而用于扩增野生型和突变型eryKR2基因的两端引物为LKR2:5'-gccatatggacgagctcgacggctggttc-3'(gc为保护性碱基,catatg为NdeⅠ酶切位点),RKR2:5'-ggaattctcaccggtcgcgcaggctctccgtc-3'(gaattc为EcoRⅠ酶切位点,前面g为保护性碱基)。
为了采用重叠PCR技术扩增TeryKR2基因,设计中间的将EryKR2酶中的PQQ突变成EryKR1酶中的LDD的正反向引物分别为TKR2-L:5'-cacgcggcgggactgctcgacgacgtcgcgatcaac gacatggac-3';TKR2-R:5'-gtcgttgatcgcgacgtcgtcgagcagtcccgccgcgtgcaccac-3';PCR扩增体系和条件参考文献[6]。
获得的PCR特异片段产物按照微量琼脂糖凝胶DNA回收试剂盒的产品说明书回收纯化后,按照文献[9],双酶切连接到载体pET28a上,再转化到大肠杆菌BL21中。从筛选到的阳性克隆中利用高纯度质粒小提试剂盒提取出重组质粒,经武汉擎科公司测序后,再利用NCBI数据库提供的核苷酸比对功能进行比对,检测质粒中克隆的基因片段与目标基因的一致性。
1.2.2 重组酮还原酶的诱导表达和酶活测定按照1%的接种量将构建的重组菌种子液接种到含100 µg/mL卡拉霉素的LB液体培养基中,37℃、150 r/min培养3 h左右加入终浓度为1 mmol/L的IPTG,继续培养4 h[10]。离心(4℃,10 000 r/min,10 min)收集菌体沉淀,用0.1 mol/L的磷酸钾缓冲液(pH7.0) 洗涤重悬后,超声波破碎(破胞强度60%,工作3 s间歇5 s,10 min)。离心(4℃,10 000 r/min,10 min)收集上清液(即粗酶液),测定其酶活、蛋白质含量及进行SDS-PAGE电泳分析。测定酶活的反应体系(3 mL):粗酶液20 μL、2 mmol/L NADPH 500 μL、2-甲基环己酮50 μL和0.1 mol/L磷酸钾缓冲液(pH7.0)。该体系置于37℃反应30 min,期间在340 nm条件下测量NADPH的吸光值变化曲线,根据曲线斜率代入NADPH标准曲线拟合的公式(y=0.004 83x+ 0.004 53,R2=0.999 82,其中y为340 nm处的吸光值,x为NADPH浓度(μmol/L)),计算NADPH含量变化情况。酶活单位U定义为在本实验条件下,1 min催化氧化1 μmol NADPH到NADP+所需要的酶量。采用Bradford法测定蛋白质含量[9],根据标准品牛血清白蛋白的浓度与吸光值的标准曲线(y=0.005 79x-0.064,R2=0.997 01,y为595 nm处的吸光值,x为蛋白质含量(µg/mL)),计算上清液中蛋白质浓度。根据文献[9]进行SDS-PAGE分析。
1.2.3 双重组菌耦合全细胞催化2-甲基环己酮的还原双重组菌耦合的发酵体系(10 mL):0.4 g E.coli BL21(pET28a-eryKR1)(或E.coli BL21(pET28a-eryKR2)、E.coli BL21(pET28a-TeryKR1)、E.coli BL21(pET28a-TeryKR2)),0.1 g E.coli BL21(pET28a-gdh),0.1 mol/L葡萄糖,0.2 mmol/L NADPH,10 mmol/L 2-甲基环己酮,0.1 mol/L磷酸钾缓冲液(pH6.0)。该体系置于30℃、120 r/min振荡反应6 h。反应液离心(4℃,10 000 r/min,10 min)收集上清并用等体积乙酸乙酯萃取,得到的上层有机相采用Agilent6890型气相色谱仪检测分析底物和产物的浓度,并按照文献[6]计算转化率、产物收率及对映体过量值(e.e值)。色谱条件为:采用毛细管柱CYCLODEXB(30 m×0.25 mm×0.25 m),载气为氮气,进样量为1 μL,FID检测器。气化温度为250℃,检测器温度为300℃;色谱柱初始温度90℃,维持4 min;升温速度20℃,终温为180℃,维持6 min;柱前压为0.27 MPa,分流比为50:1。
2 结果 2.1 重组质粒的构建及鉴定eryKR1基因和eryKR2基因的突变位点两侧的PCR扩增产物TeryKR1-L、TeryKR1-R、TeryKR2-L和TeryKR2-R分别进行1.0%琼脂糖凝胶电泳检测,结果如图 1和图 2,可见TeryKR1-L和TeryKR2-L均位于1 000 bp附近,与两个目标基因突变位点左侧的序列长度分别为965 bp和893 bp吻合,而片段TeryKR1-R和TeryKR2-R都略高于500 bp,与两个目标基因的突变位点右侧的序列长度分别为550 bp和553 bp吻合。

|
| 图 1 eryKR1基因的突变位点两侧的PCR扩增产物的1.0%琼脂糖凝胶电泳图 M:DL5000 DNA ladder;1:TeryKR1-L的PCR扩增产物;2:TeryKR1-R的PCR扩增产物 |

|
| 图 2 eryKR2基因的突变位点两侧的PCR扩增产物的1.0%琼脂糖凝胶电泳图 M:DL5000 DNA ladder;1:TeryKR2-L的PCR扩增产物;2:TeryKR2-R的PCR扩增产物 |
突变型eryKR1基因(命名为TeryKR1)和突变型eryKR2基因(命名为TeryKR2)的重叠PCR产物,野生型eryKR1基因和eryKR2基因的PCR扩增产物,分别以1.0 %琼脂糖凝胶进行电泳检测,结果如图 3-图 6。产物片段的大小都位于1 000 bp和2 000 bp之间,与目标eryKR1基因和eryKR2基因的序列长度分别为1 461 bp和1 395 bp吻合。

|
| 图 3 突变型eryKR1基因(TeryKR1)的PCR产物的1.0%琼脂糖凝胶电泳图 M:DL5000 DNA ladder;1:TeryKR1的PCR扩增产物 |

|
| 图 4 突变型eryKR2基因(TeryKR2)的PCR产物的1.0%琼脂糖凝胶电泳图 M:DL5000 DNA ladder;1:TeryKR2的PCR扩增产物 |

|
| 图 5 野生型eryKR1基因的PCR产物的1.0%琼脂糖凝胶电泳图 M:DL5000 DNA ladder;1:eryKR1的PCR扩增产物 |

|
| 图 6 野生型eryKR2基因的PCR产物的1.0%琼脂糖凝胶电泳图 M:DL5000 DNA ladder;1:eryKR2的PCR扩增产物 |
将基因片段eryKR1、eryKR2、TeryKR1和Tery-KR2分别克隆到pET28a载体上,构建了重组质粒pET28a-eryKR1、pET28a-eryKR2、pET28a-TeryKR1和pET28a-TeryKR2。对获得的重组质粒分别进行双酶切鉴定(图 7),证实了4个重组质粒中克隆的基因片段均在1 000和2 000 bp之间,与目标片段的大小一致。含有这4个质粒的重组大肠杆菌分别命名为E.coli BL21(pET28a-eryKR1)、E.coli BL21(pET28a-eryKR2)、E.coli BL21(pET28a-TeryKR1)和E.coli BL21(pET28a-TeryKR2)。

|
| 图 7 重组质粒双酶切鉴定产物1.0%琼脂糖凝胶电泳图 M:DL5000 DNA ladder;1:重组质粒pET28a-eryKR1的双酶切鉴定产物;2:重组质粒pET28a-TeryKR1的双酶切鉴定产物;3:重组质粒pET28a-eryKR2的双酶切鉴定产物;4:重组质粒pET28a-TeryKR2的双酶切鉴定产物 |
利用NCBI数据库提供的核苷酸比对功能,对重组质粒的测序结果进行序列分析,得出测序结果中克隆的野生型和突变型eryKR1基因、野生型和突变型eryKR2基因与登录号为AM420293.1报道的糖多孢红霉菌NRRL2338的基因组DNA中792 285-793 727 bp、796 692-798 068 bp的碱基序列的相似度分别达到100%、99%、100%和99%。
突变型eryKR1和eryKR2基因序列的核苷酸比对结果分别如图 8和图 9,突变型eryKR1基因中对应AM420293.1序列中的第793 209-793 218个碱基ctcgacgac(编码的氨基酸残基为LDD)已替换成测序结果中的ccgcagcag(编码的氨基酸残基为PQQ);而突变型eryKR2基因中对应AM420293.1序列中的第79 7547-797 556个碱基ccgcagcag(编码的氨基酸序列为PQQ)已替换成测序结果中的ctcgacgac(编码的氨基酸序列为LDD),说明质粒pET28a-TeryKR1和pET28a-TeryKR2中克隆的突变型eryKR1和eryKR2基因编码的氨基酸序列,其中调控底物的立体选择性还原的氨基酸残基发生了LDD↔PQQ互换。

|
| 图 8 重组质粒pET28a-TeryKR1的核苷酸比对结果(截图) Query:重组质粒pET28a-TeryKR1的测序结果(红色实线标出的为突变位点);Sbjct:糖多孢红霉菌NRRL2338的基因组DNA中对应位点 |

|
| 图 9 重组质粒pET28a-TeryKR2的核苷酸比对结果(截图) Query:重组质粒pET28a-TeryKR2的测序结果(红色实线标出的为突变位点);Sbjct:糖多孢红霉菌NRRL2338的基因组DNA中对应位点 |
以E.coli BL21(pET28a)菌为阴性对照,SDS-PAGE检测经IPTG诱导后重组菌E.coli BL21(pET-28a-eryKR1)、E.coli BL21(pET28a-TeryKR1)、E.coli BL21(pET28a-eryKR2)和E.coli BL21(pET28a-Ter-yKR2) 中目标蛋白的表达情况。如图 10和11所示,与阴性对照中的蛋白质条带不同,经IPTG诱导后的重组菌在45.0 kD和66.2 kD之间都有酮还原酶的表达条带(目标蛋白的相对分子量为57 kD,包括了pET28a载体表达的His tag片段)。

|
| 图 10 野生型和突变型酮还原酶EryKR1的SDS-PAGE检测图 M:标准蛋白Marker;1-3:经IPTG诱导的E.coli BL21(pET28a-eryKR1);4,5:经IPTG诱导的E.coli BL21(pET28a-TeryKR1);6:经IPTG诱导的阴性对照E.coli BL21(pET28a) |

|
| 图 11 野生型和突变型酮还原酶EryKR2的SDS-PAGE检测图 M:标准蛋白Marker;1-3:经IPTG诱导的E.coli BL21(pET28a-eryKR2);4-6:经IPTG诱导的E.coli BL21(pET28a-TeryKR2);7:经IPTG诱导的阴性对照E.coli BL21(pET28a) |
利用BandScan 5.0软件分析这些重组菌的总可溶性蛋白的SDS-PAGE图,可知,重组菌E.coli BL21(pET28a-TeryKR1)中目标蛋白质的表达量占全菌可溶性蛋白质的8.49%(图 10中的孔道4),E.coli BL21(pET28a-eryKR1)中eryKR1的表达量为5.66%(图 10中孔道2),而重组菌E.coli BL21(pET28a-eryKR2)(图 11中孔道3) 和E.coli BL21(pET28a-TeryKR2)(图 11中孔道5) 中表达出来的eryKR2分别占全菌可溶性蛋白质的5.92%和5.04%。取这些孔道对应的重组菌进行后续的酶活和生物催化2-甲基环己酮不对称还原实验。
2.3 野生型和突变型酮还原酶的活性E.coli BL21(pET28a-eryKR1)、E.coli BL21(pET28a-TeryKR1)、E.coli BL21(pET28a-eryKR2)和E.coli BL21(pET28a-TeryKR2)的重组酮还原酶的粗酶液酶活分别为187.02 U/L、192.94 U/L、51.02 U/L和35.43 U/L,其中的蛋白质浓度分别为125.73 μg/mL、205.52 μg/mL、138.26 μg/mL和115.68 μg/mL,由此得出粗酶液的比酶活分别为1.49 U/mg、0.94 U/mg、0.37 U/mg和0.31 U/mg。根据野生型或突变型酮还原酶粗酶液的酶活和比酶活数据,可知控制立体选择性还原的位点(motif)处的氨基酸序列LDD突变成PQQ(LDD→PQQ)的突变型eryKR1的比酶活,比起野生型eryKR1的略微有所下降,这说明突变对酶的活性有所影响。另外,与eryKR1相比,eryKR2当以2-甲基环己酮作为底物时,酶活较低,而PQQ→LDD的突变型eryKR2与野生型eryKR2的比酶活相较,变化不大。
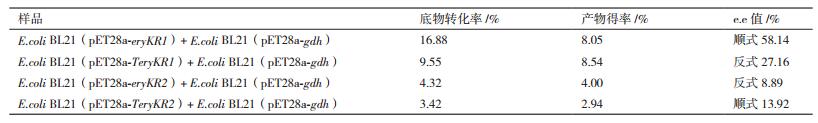
2.4 双重组菌耦合催化2-甲基环己酮不对称还原实验气相色谱检测构建的四个重组大肠杆菌分别与E.coli BL21(pET28a-gdh)耦合还原2-甲基环己酮的转化液(图 12),根据底物和产物的峰面积,利用外标法计算底物及产物的浓度,进而得出转化率、产物产率及对映体过量值(e.e值),数据见表 1,可知:与阴性对照的转化液中没有还原产物检出不同(图 12-E),野生型eryKR1催化2-甲基环己酮不对称还原的产物以顺式2-甲基环己醇为主,产率为8.05%,e.e值为58.14%(图 12-A),而突变型eryKR1催化不对称还原的产物主要为反式,产率为8.54%,e.e值为27.16%(图 12-B);野生型eryKR2催化2-甲基环己酮不对称还原,产物以反式2-甲基环己醇为主,产率仅为4.00%,e.e值为8.89%(图 12-C),而突变型eryKR2催化不对称还原的产物主要为顺式,产率2.94%,e.e值为13.92%(图 12-D),说明eryKR1和eryKR2中分别发生LDD→PQQ和PQQ→LDD的突变时,催化2-甲基环己酮不对称还原生成的主要产物的立体结构发生改变,这扩展了LDD模式序列为酮还原酶中控制链式聚酮链立体选择性还原的位点的理论,得出该位点也是控制2-甲基环己酮不对称还原的模序。

|
|
图 12 双重组菌耦合还原2-甲基环己酮的转化液的气相色谱检测图谱
A : E.coli BL21(pET28a-eryKR1)+ E.coli BL21(pET28a-gdh);B : E.coli BL21(pET28a-TeryKR1)+ E.coli BL21(pET28a-gdh); C : E.coli BL21(pET28a-eryKR2)+ E.coli BL21(pET28a-gdh);D : E.coli BL21(pET28a-TeryKR2)+ E.coli BL21(pET28a-gdh); E : E.coli BL21(pET28a)+ E.coli BL21(pET28a-gdh) |
另外,根据酮还原酶催化链式聚酮链立体选择性还原的情况,eryKR1为B型酮还原酶,而eryKR2为A型KR,本研究结果还扩展了该理论,即eryKR1和eryKR2催化含环己基结构的化合物还原时形成的主要产物的立体结构也是不同的。
3 讨论对聚酮合成酶的特殊结构、催化机制的研究,有助于了解酶的催化、分子识别及蛋白质与蛋白质相互作用的分子机制[11],其中聚酮合成酶中的酮还原酶如何控制底物的立体选择性还原的机制一直是研究的热点。Ⅰ型聚酮合成酶的酮还原酶域(ketoreductase,KR)根据链式聚酮链立体选择性还原的情况分为A型和B型,这两种KR在第88-103和第134-149个氨基酸处的两个模式序列(motif)有很大区别。B型KR如EryKR1,在第88-10个氨基酸处有一个LDD模式序列,而A型KR如EryKR2,没有这个模式。另外,在第134-149个氨基酸处,B型KR有个P144、N148,而A型KR中是W141。若这些位点发生了突变,酶域对(2R,S)-2-甲基-3-羰基戊酰N-乙酰半胱酰胺硫酸酯的立体选择性会发生相应的改变,证实了这些酶域中存在有控制立体选择性还原的模式序列[4, 7, 8]。另外,A型和B型KR的晶体结构分析显示A型KR中的W残基与B型KR中的LDD序列分别位于这两种KR的活性中心裂缝的两侧,说明这两种类型的KR中底物是从不同的方向被引至酶的活性中心,导致底物中的β-羰基以不同方向结合NADPH,从而生成不同立体结构的产物[12]。而且,与B型KR不同,A型KR的活性中心处于一种井然有序、随时准备好催化的Lys-2’-O-核糖-Tyr的氢键结合的状态。在底物和辅酶结合时,A型KR会关闭盖子环(lid loop),限制底物只从一个方向进入活性中心,导致生成的产物具有特定的立体结构[13]。在这个过程中,酶的一些残基能够有助于底物的定位[14]。
聚酮合成酶的酮还原酶域可以还原的底物类型多种多样,不仅能对类似天然底物——链式聚酮链的羰基底物有较好的还原能力,如(2R,S)-2-甲基-3-羰基戊酰N-乙酰半胱酰胺硫酯[3],而且还能以那些结构不同于链式聚酮链的化合物作为底物,如(9RS)-反式-1-萘烷酮[3]、脂环酮[5]等。但是,目前针对聚酮合成酶酮还原酶域的控制立体选择性还原的机理研究,多以(2R,S)-2-甲基-3-羰基戊酰N-乙酰半胱酰胺硫酸酯、(9RS)-反式-1-萘烷酮为研究底物,对聚酮合成酶的酮还原酶域为何能结合潜手性取代环己酮以及如何控制此反应的立体选择性机理还未见报道。本研究基于LDD模式序列可能也负责控制取代环己酮类底物的立体选择性还原的假设,构建了分别异源表达聚酮合成酶模块1的酮还原酶(EryKR1)、模块2的酮还原酶(EryKR2)、LDD残基替换为PQQ(LDD→PQQ)的EryKR1及PQQ替换为LDD(PQQ→LDD)的EryKR2的重组大肠杆菌,考察了野生型模块1和模块2的重组酮还原酶对2-甲基环己酮的不对称还原能力,结果显示在底物浓度为10 mmol/L的条件下,EryKR1的还原产物主要为顺式-2-甲基环己醇,产率为8.05%,e.e值为58.14%,而EryKR2的还原产物主要为反式-2-甲基环己醇,产率为4.00%,e.e值为8.89%,说明即使以2-甲基环己酮为底物,模块1和模块2的酮还原酶的立体选择性还原情况也是不同的。通过比较突变前后的重组EryKR1和EryKR2以2-甲基环己酮为底物的酶活及产物的立体结构,结果发现虽然突变对酶活的影响不大,但是突变后的重组EryKR1催化还原2-甲基环己酮的主要产物由突变前的顺式-2-甲基环己醇转变成反式-2-甲基环己醇,而突变后的重组EryKR2催化的主要还原产物的立体结构则由突变前的反式变成了顺式,说明酮还原酶催化2-甲基环己酮还原的主要产物的立体结构变化与酶中LDD模式序列的突变之间存在关联,得出LDD模序确实为酶中控制取代环己酮类底物立体选择性还原的位点。
4 结论本研究构建了分别异源表达聚酮合成酶模块1的酮还原酶(EryKR1)、模块2的酮还原酶(EryKR2)、LDD残基替换为PQQ(LDD→PQQ)的EryKR1和PQQ替换为LDD(PQQ→LDD)的EryKR2的重组大肠杆菌,比较突变型和野生型重组大肠杆菌还原2-甲基环己酮的主要产物的立体结构,得出LDD→PQQ的突变使得EryKR1的主要还原产物从顺式-2-甲基环己醇转变成反式-2-甲基环己醇,而PQQ→LDD的突变可使EryKR2的主要还原产物2-甲基环己醇的立体结构由反式变成顺式,确定了聚酮合成酶的酮还原酶的LDD模式序列为控制2-甲基环己酮立体选择性还原的位点。
| [1] | Long JX, Shu SY, Wu QY, et al. Selective cyclohexanol production from the renewable lignin derived phenolic chemicals catalyzed by Ni/MgO[J]. Energy Conversion and Management, 2015, 105 (15): 570–577. |
| [2] | Conti RMD, Porto ALM, Augusto J, et al. Microbial reduction of cyclohexanones[J]. Journal of Molecular Catalysis B:Enzymatic, 2001, 11 (4-6): 233–236. DOI:10.1016/S1381-1177(00)00087-4 |
| [3] | Siskos AP, Baerga-Ortiz A, Bali S, et al. Molecular basis of Celmer's rules:stereochemistry of catalysis by isolated ketoreductase domains from modular polyketide synthases[J]. Chemistry & Biology, 2005, 12 (10): 1145–1153. |
| [4] | Piasecki SK, Taylor CA, Detelich JF, et al. Employing modular polyketide synthase ketoreductases as biocatalysts in the preparative chemoenzymatic syntheses of diketide chiral building blocks[J]. Chemistry & Biology, 2011, 18 (10): 1331–1340. |
| [5] | Bali S, O'Hare HM, Weissman KJ. Broad substrate specificity of ketoreductases derived from modular polyketide synthases[J]. ChemBiochem, 2006, 7 (3): 478–484. DOI:10.1002/cbic.v7:3 |
| [6] | 李凌凌, 吕早生, 关海燕, 等. 糖多孢红霉菌酮还原酶域在羰基还原中的应用[J]. 华中科技大学:自然科学版, 2011, 39(2): 72–75. |
| [7] | Baerga-Ortiz A, Popovic B, Siskos AP, et al. Directed mutagenesis alters the stereochemistry of catalysis by isolated ketoreductase domains from the erythromycin polyketide synthase[J]. Chemistry & Biology, 2006, 13 (3): 277–285. |
| [8] | O'Hare HM, Baerga-Ortiz A, Popovic B, et al. High-throughput mutagenesis to evaluate models of stereochemical control in ketoreductase domains from the erythromycin polyketide synthase[J]. Chemistry & Biology, 2006, 13 (3): 287–296. |
| [9] | 杨忠华, 左振宇, 黄皓, 等. 生物工程专业实验[M]. 北京: 化学工业出版社, 2014: 65-73. |
| [10] | 李凌凌, 吕早生, 左振宇, 等. 嗜酸喜温硫杆菌硫加氧还原酶基因的克隆、表达及酶活性研究[J]. 生物技术通报, 2015, 31(11): 186–194. |
| [11] | Shen B. Polyketide biosynthesis beyond the typeⅠ, Ⅱand Ш polyketide synthase paradigms[J]. Current Opinion in Chemical Biology, 2003, 7 (2): 285–295. DOI:10.1016/S1367-5931(03)00020-6 |
| [12] | Keatinge-Clay AT. A tylosin ketoreductase reveals how chirality is determined in polyketides[J]. Chemistry & Biology, 2007, 14 (8): 898–908. |
| [13] | Bonnet SA, Whicher JR, Papireddy K, et al. Structural and stereochemical analysis of a modular polyketide synthase ketoreductase domain required for the generation of a cis-alkene[J]. Chemistry & Biology, 2013, 20 (6): 772–783. |
| [14] | Zheng J, Taylor CA, Piasecki SK, et al. Structural and functional analysis of A-type ketoreductases from the amphotericin modular polyketide synthase[J]. Structure, 2010, 18 (8): 913–922. DOI:10.1016/j.str.2010.04.015 |