




2. 江南大学工业生物技术教育部重点实验室,无锡 214122
2. Key Laboratory of Industrial Biotechnology of Ministry of Education, Jiangnan University, Wuxi 214122
乳酸克鲁维酵母(Kluyveromyces lactis)是美国FDA(Food and Drug Administration)认定为食品安全级(Generally regarded as safe,GRAS)的酵母菌,具有碳源利用范围广、生长速率快、能够分泌高分子量蛋白、不会对蛋白高度糖基化以及不产生内毒素等优势[1]。同时,与甲醇营养型酵母相比,K. lactis在发酵过程不需要添加有毒性的诱导物及防爆设备[2],并且分泌外源蛋白的能力高于S. cerevisiae[3],在食品和医药领域已实现规模化生产及应用[4]。此外,2004年Dujon等[5]完成了K. lactis的全基因组测序,有利于深入了解并合理改造该菌株。然而,K. lactis在外源蛋白分泌表达中仍存在瓶颈,其蛋白产量还有较大的提升空间。目前,很多研究者在发酵条件优化,载体系统包括高效信号肽、强启动子、筛选标记和整合方式等的选择,宿主菌的工程改造包括糖基化基因的敲除、分子伴侣的共表达等方面进行了探索,以期获得高效分泌表达外源蛋白的工程菌株[1, 6]。
启动子是位于结构基因5'端上游的DNA序列,一般由100-1 000个碱基对构成,是RNA聚合酶和转录因子的结合位点,行使起始基因转录的功能[7, 8]。作为基因表达调控的顺式元件,启动子在转录水平调控中占据关键的地位,其活性高低在很大程度上影响基因的表达水平,进而影响代谢途径的效率以及外源蛋白的产量[9]。目前,一些学者在实验室水平利用个别启动子进行了初步的应用研究,但是在K. lactis中针对启动子的系统理论研究鲜有报道[6],用于调控外源基因表达的启动子选择少,缺乏对不同启动子间功能比较的研究,限制了进一步挖掘功能良好的启动子元件。因此,评价不同启动子在K. lactis中的转录功能,对于高效表达外源蛋白具有重要的意义。
通常利用报告基因的表达量来评价启动子的转录活性及功能,常用的报告基因包括β-葡萄糖苷酸酶基因(gus)、氯霉素乙肽转移酶基因(cat)、荧光素酶基因(luc)和绿色荧光蛋白基因(gfp)。GFP是从水母(Aequorea victoria)体内克隆得到的一种荧光蛋白,经紫外光和蓝光激发无需辅助因子及底物即可发射稳定的绿色荧光,具有检测方便、活体表达、对细胞无毒害、无种族和组织特异性等优点,在分子生物学、生物传感器、细胞生物学以及药物研究中得到广泛的应用[10, 11]。通过对gfp基因进行突变改造,获得增强型绿色荧光蛋白(EGFP),与GFP相比其蛋白合成、折叠效率及荧光强度更高[12]。
为了发挥K. lactis作为宿主菌株表达外源蛋白的优势,基于当前针对K. lactis中不同启动子功能比较的研究相对匮乏的现状,亟待开展启动子功能的研究来寻找具有高效启动外源蛋白表达能力的启动子。本研究以egfp为报告基因,初步研究乙醇脱氢酶启动子(padh)、3-磷酸甘油脱氢酶启动子(pgpd)、麦芽糖酶启动子(pmal)、半乳糖苷酶启动子(pgal)在K. lactis中的转录功能及在相应诱导条件下[13-18]转录活性的变化。以此为基础,为实现在K. lactis中分析和比较不同启动子的功能提供参考。
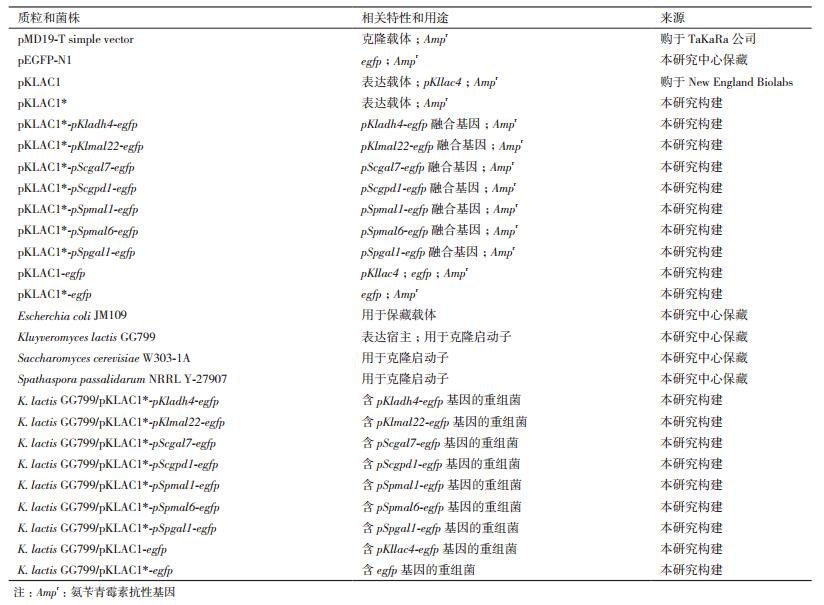
1 材料与方法 1.1 材料 1.1.1 菌株和质粒实验所用的质粒和菌株见表 1。
采用启动子在线测评软件,对NCBI数据库报道的相关序列的开放阅读框上游核苷酸序列进行在线预测,利用oligo7设计PCR特异性引物(表 2),由金唯智公司合成。
LB培养基(g/L):蛋白胨10.0,酵母粉5.0,NaCl 10.0,固体培养基添加20 g/L琼脂粉,添加终浓度为100 μg/mL的氨苄青霉素用于大肠杆菌(Escherchia coli)转化子的筛选;YEPD培养基(g/L):葡萄糖20.0,蛋白胨20.0,酵母粉10.0,用于K. lactis、S. cerevisiae及S. passalidarum的培养;YCB固体培养基(100 mL):酵母基础碳源(YCB)1.17 g,pH 7.0的1 mol/L KH2PO4-K2HPO4缓冲液3 mL(终浓度30 mmol/L),琼脂粉2 g,灭菌后加入乙酰胺至终浓度5 mmol/L,用于K. lactis重组菌的筛选;诱导培养基:YEPD培养基添加6% NaCl或5 g/L乙醇;YEPDG培养基(g/L):葡萄糖10.0,半乳糖10.0,蛋白胨20.0,酵母粉10.0;YEPG培养基(g/L):半乳糖20.0,蛋白胨20.0,酵母粉10.0;YEPDM培养基(g/L):葡萄糖10.0,麦芽糖10.0,蛋白胨20.0,酵母粉10.0;YEPM培养基(g/L):麦芽糖20.0,蛋白胨20.0,酵母粉10.0。
1.1.4 主要试剂和仪器Hind Ⅲ、Bgl Ⅱ、Sal Ⅰ、Bst Ⅺ等限制性内切酶(美国Fermentas公司);T4 DNA连接酶、PrimeSTAR、Sac Ⅱ、Dpn Ⅰ(大连TaKaRa公司);质粒小量提取试剂盒、DNA片段纯化试剂盒、胶回收试剂盒、2×Pfu PCR Master Mix、2×Taq PCR Master Mix(杭州宝赛生物技术有限公司);氨苄青霉素购自上海生工公司;Yeast Carbon Base购自New England Biolabs公司;其他试剂为国产分析纯。Bio-Rad S1000 PCR仪、Chemi Doc凝胶成像仪(美国Bio-Rad公司);Multiporator电转仪(德国Eppendorf公司);荧光显微镜ECLIPSE 50i(日本NiKon公司);荧光分光光度计F-7000(日本日立公司)。
1.2 方法 1.2.1 缺失Kllac4启动子功能的载体pKLAC1*的构建参照Madhavan等[19]的方法,以载体pKLAC1为模板,采用Oligo 7设计引物KlacF/KlacR,PCR扩增得到线性化的载体片段。PCR反应体系(100 µL):PrimeSTAR 0.5 µL,5×Buffer 20 µL,dNTP 8 µL,模板1 µL,上下游引物(25 µmol/L)各1 µL,灭菌的ddH2O 68.5 µL。PCR扩增条件为:98℃ 2 min;98℃ 20 s,55℃ 20 s,72℃ 8 min,30个循环;72℃ 5 min。纯化PCR产物,Dpn Ⅰ 37℃消化反应2 h后,经Hind Ⅲ酶切后纯化,16℃条件下T4 DNA连接酶将线性化的载体片段自连形成环状质粒,转化E. coli JM109,涂布Amp抗性平板,挑取阳性转化子分别经Bst Ⅺ、Sac Ⅱ、Hind Ⅲ酶切验证正确后,送上海生工公司测序,测序正确的载体命名为pKLAC1*。
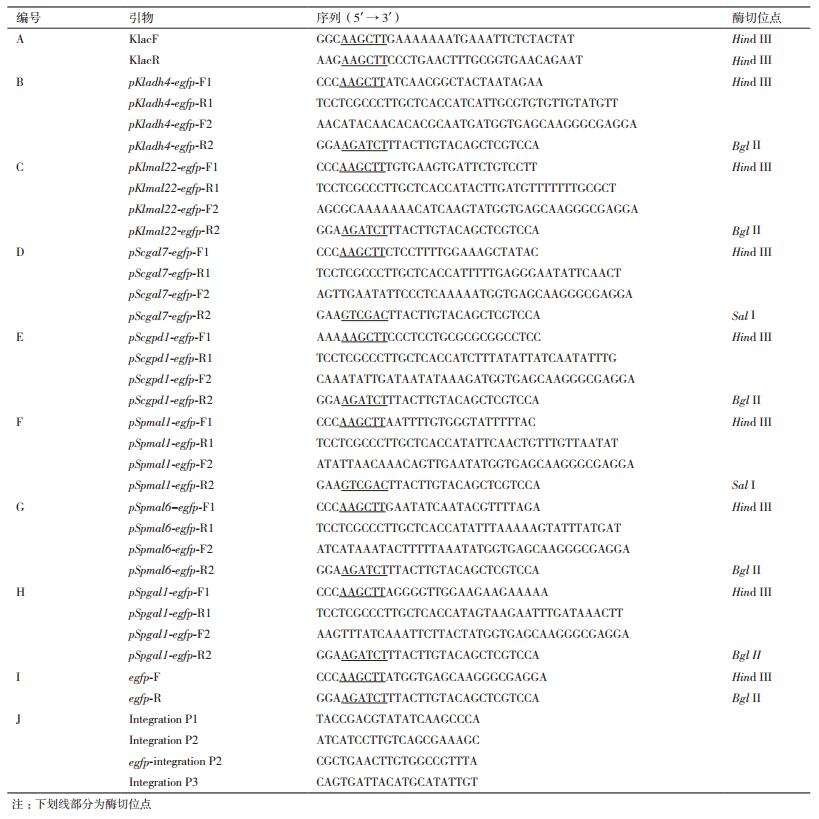
1.2.2 启动子与报告基因egfp的克隆及重叠延伸PCR根据已报道的来源于K. lactis的adh4、mal22启动子、S. cerevisiae的gal7、gpd1启动子、S. passalidarum的mal1、mal6、gal1启动子基因序列以及质粒pEGFP-N1中egfp序列,采用Oligo 7设计重叠延伸PCR引物(表 2)。用酸性玻璃珠法分别提取K. lactis、S. cerevisiae、S. passalidarum染色体基因组,并以基因组和质粒pEGFP-N1为模板,用表 2中相应的引物重叠延伸PCR扩增得到promoter-egfp融合片段。将PCR产物纯化后TA克隆,转化E. coli JM109,筛选阳性转化子送上海生工公司测序。
1.2.3 重组表达载体的构建将测序结果正确的promoter-egfp片段酶切后插入载体pKLAC1*,egfp片段酶切后分别插入载体pKLAC1和pKLAC1*,转化E. coli JM109,提取质粒酶切验证,构建成功的重组表达载体命名为pKLAC1*-promoter-egfp、pKLAC1-egfp、pKLAC1*-egfp。
1.2.4 重组菌的构建及单拷贝整合重组菌的筛选Bst Ⅺ线性化重组表达载体后电转化K. lactis GG799,涂布YCB平板,30℃培养4 d,挑选转化子于YCB平板纯化多次,接种于YEPD培养基,提取重组菌染色体基因组。参照Read等[20]的方法略作改进,线性化的同源重组片段在染色体中lac4位点的启动子区域发生双交换整合,设计特异性引物(表 2),分别利用单拷贝整合引物Integration P1/ egfp integration P2(Integration P2) 和多拷贝整合引物egfp integration P2(Integration P2)/Integration P3引物进行PCR扩增验证重组菌的整合拷贝数。为了在同一水平上比较不同启动子的功能,挑选单拷贝整合重组菌(各挑选3个)进行EGFP表达量的分析。
1.2.5 单拷贝整合重组菌EGFP的观察YEPD平板活化重组菌,挑取单菌落分别接种于相应培养基(表 3),30℃,200 r/min,培养36 h。收集菌体细胞并用生理盐水洗涤2次,吸取少量菌悬液置于载玻片上,在Nikon ECLIPSE 50i荧光显微镜下观察菌体细胞并拍照,放大倍数为100倍,激发波长λ=485 nm,探究不同启动子在K. lactis中的转录功能及诱导前后启动强度的变化。

YEPD平板活化重组菌,挑取单菌落分别接种于相应培养基(表 3),30℃,200 r/min,培养36 h。收集菌体细胞并用生理盐水洗涤2次后将菌体重悬至OD600为0.5左右,用荧光分光光度计F-7000对菌体胞内EGFP的荧光强度定量测定,每个菌株在测定时设置3个平行。主要设置参数为:Wavelength:Excitation=488 nm,Emission=510 nm;EX 5.0 nm,EM 5.0 nm,PMT Voltage 400 V。
2 结果 2.1 载体pKLAC1*的构建通过PCR方法使载体pKLAC1自身Kllac4启动子区域起始密码子上游700 bp左右的序列缺失,破坏其3个TATA框及UAS元件,使pKllac4无法起始转录,得重组表达载体pKLAC1*(图 1-A),以此为基础评价不同启动子的功能。如图 1-B所示,在7 500-10 000 bp有单一扩增条带,与目的片段大小(8 438 bp)一致,酶切验证及测序结果表明成功构建载体pKLAC1*。

|
| 图 1 重组表达载体pKLAC1*的构建(A)及酶切(B)验证 (A)载体pKLAC1*的构建图谱。(B)M:DL 15 000 Marker;1:KlacF/KlacR引物PCR扩增产物;2:pKLAC1* Bst XI单酶切;3:pKLAC1* Sac Ⅱ单酶切;4:pKLAC1* Hind Ⅲ单酶切 |
根据1.2.3所述方法得重组载体pKLAC1*-promoter-egfp,以pKLAC1-egfp和pKLAC1*-egfp分别为阳性(PC)和阴性(NC)对照(图 2)。以pKladh4为例,PCR扩增启动子片段(1 205 bp)、egfp片段(720 bp)及重叠延伸PCR扩增融合片段(1 925 bp)的核酸电泳如图 3-A所示,Hind Ⅲ与Bgl Ⅱ双酶切得到8.4 kb和2.0 kb两个片段,与目的基因大小一致,表明重组载体构建成功。PC质粒经Hind Ⅲ和Bgl Ⅱ双酶切得到8.1 kb和0.7 kb两个片段(图 3-B),NC质粒经Hind Ⅲ和Bgl Ⅱ酶切得到8.8 kb和0.7 kb两个片段(图 3-C)。

|
| 图 2 重组载体pKLAC1*-promoter-egfp(A)、pKLAC1-egfp(B)及pKLAC1*-egfp(C)的构建图谱 |

|
| 图 3 目的基因克隆及重组表达载体的酶切验证 (A)1:pKladh4片段;2:egfp片段;3:pKladh4-egfp融合片段;4:载体pKLAC1*-pKladh4-egfp Hind Ⅲ和Bgl Ⅱ双酶切。(B)1:egfp片段;2:载体pKLAC1-egfp Hind Ⅲ和Bgl Ⅱ双酶切。(C)1:egfp片段;2:载体pKLAC1*-egfp Hind Ⅲ和Bgl Ⅱ双酶切;M:λ DNA/Pst Ⅰ Marker |
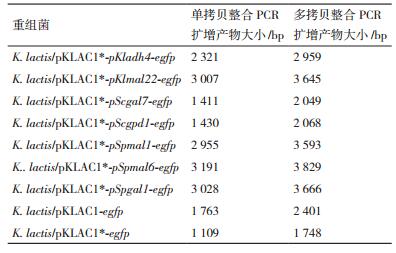
构建成功的重组载体经Bst XI酶切线性化后分别电转化K. lactis GG799,得表 1所示的9个重组菌。同源重组片段在lac4位点处发生单拷贝整合或多拷贝串联整合(图 4-A),利用单拷贝整合引物PCR扩增得到相应大小的片段,表明重组片段成功整合入K. lactis染色体并发生单拷贝整合;利用多拷贝整合引物PCR扩增得到相应大小的片段,表明重组片段发生多拷贝整合。含不同整合拷贝数的重组菌PCR扩增相应的产物(表 4) 经琼脂糖凝胶电泳分析(图 4-B),挑选单拷贝整合重组菌进行后续实验。

|
| 图 4 重组菌整合拷贝数的验证 (A)同源重组片段在K. lactis染色体lac4位点启动子区域发生不同拷贝数整合原理图。(B)K. lactis/pKLAC1*-pKladh4-egfp:1,3,5:单拷贝整合引物PCR产物;2,4,6:多拷贝整合引物PCR产物;M:λ DNA/Pst Ⅰ Marker(1、2表明单拷贝重组菌;3、4表明多拷贝重组菌;5、6为阴性对照) |
为了在同一水平上比较不同启动子在K. lactis中的功能,在上述重组菌中筛选单拷贝整合的重组菌,因切除α因子信号肽,EGFP在胞内表达,通过荧光显微镜观察各重组菌EGFP的表达情况。如图 5所示,pKladh4、pKlmal22、pScgal7、pScgpd1在K.lactis中实现了EGFP的有效表达,并且相对于YPD培养条件下,加入相应诱导物后其表达能力有一定的提高。同时,来源于S. cerevisiae的启动子转录活性优于来源于自身的启动子,表明pScgal7、pScgpd1具有较强的起始转录外源基因的能力。来源于S. passalidarum的pSpmal1、pSpmal6和pSpgal1表达绿色荧光蛋白能力较弱。

|
| 图 5 不同启动子调控下单拷贝整合重组菌胞内EGFP的观察(100×) |
根据1.2.5所述方法检测重组菌胞内EGFP的荧光强度,评价不同启动子起始转录能力的强弱以及诱导前后强度的变化,结果如图 6所示。来源于K. lactis的pKladh4受乙醇诱导,分别在YEPD以及添加5 g/L乙醇后的培养基中测定荧光强度。pKladh4活性略高于作为阳性对照的pKllac4启动子,在YEPD培养条件下,前者启动子强度为后者的1.3倍,转录外源基因能力较强。相同的是,两者在YEPD中即存在较强的本底表达,在添加相应的诱导物后,启动子强度略有提高,两者表达量均为在YEPD培养基中表达量的1.13倍。pKlmal22受麦芽糖诱导,在YEPD中强度较弱,在YEPM和YEPDM培养基中绿色荧光蛋白的表达量分别提高了46%和15%。

|
| 图 6 不同启动子调控下单拷贝整合重组菌荧光强度的定量测定 |
S. cerevisiae来源的pScgal7和pScgpd1具有较强的活性,其荧光强度分别是阳性对照的1.37倍和2.22倍。其中pScgal7在YEPG条件下EGFP的表达量提高了57%,而在YEPDG中荧光强度相对于YEPG培养基中略有降低。pScgpd1受渗透压调控,在YEPD中添加6% NaCl后,启动子活性提高了22%。
来源于S. passalidarum的pSpmal1、pSpmal6和pSpgal1启动强度弱,EGFP表达极弱,其中pSpmal1和pSpgal1分别在麦芽糖和半乳糖诱导下活性最高提高了31%和12%,但仍处于较低水平。
3 讨论K. lactis作为表达宿主具有多种优势已实现上百种外源蛋白的成功表达。K. lactis有两种类型的表达系统,一是游离型表达载体(如pKARS,pKD1),二是整合型表达载体(如pKLAC1)[6, 21]。后者将外源基因整合于基因组中lac4位点并受pKllac4调控,α因子信号肽介导下将外源蛋白分泌到胞外,因K. lactis分泌杂蛋白较少,大大降低了后续分离纯化的成本[4, 22]。目前,外源蛋白表达量仍限制于相对较低的水平。研究表明,外源蛋白调控水平包括转录水平、翻译水平和翻译后修饰水平[23],目的蛋白的性质、宿主细胞及其发酵条件、表达载体系统、启动子的选择、密码子的使用、信号肽、翻译信号、蛋白折叠以及分泌等因素都会影响外源蛋白的分泌表达[1],随着现代分子生物学技术的发展,有助于人们进一步探索各表达元件、分子伴侣等对外源蛋白分泌表达的影响。
启动子活性高低在很大程度上影响外源蛋白的表达,因此,研究启动子成为调控外源蛋白常用的一种方法。目前,已报道来源于K. lactis的启动子只有很少一部分用于调控外源蛋白的表达。最常用的启动子包括S. cerevisiae来源的组成型磷酸甘油酸激酶基因(pgk)启动子和诱导型酸性磷酸酶基因(pho5)启动子[24, 25],K. lactis来源的β-半乳糖苷酶基因(lac4)启动子。在培养基中分别添加磷酸盐、半乳糖或乳糖诱导这些启动子调控外源蛋白的表达,然而,在缺乏诱导物的情况下其转录活性并未受到完全抑制。当前,对启动子的研究主要着眼于表达某种特定的外源蛋白,存在一定的特异性和局限性,针对同一评价体系中不同启动子之间的功能差异的比较鲜有报道。因此,分析不同启动子功能、筛选性能优越的启动子对于时空调控外源蛋白表达及代谢途径控制起着至关重要的作用。
本研究对所选择的启动子活性差异作了初步探索,表明该体系的可行性,基于该评价体系对启动子转录活性的研究,为挖掘性能优越的启动子调控外源蛋白的高效表达提供参考。荧光检测结果表明,pKladh4、pScgal7、pScgpd1均能高效启动EGFP在K. lactis中的表达,且在相应的诱导条件下启动能力有所增强,EGFP的表达量均高于当前应用最广泛的Kllac4启动子。pKladh4具有较强的本底表达能力,Saliola等[13]证实与酿酒酵母不同,K. lactis中乙醇脱氢酶在葡萄糖和乙醇同时存在条件下可表达,不存在葡萄糖阻遏效应,因此在培养基加入乙醇后活性提高。在S. cerevisiae中,半乳糖苷酶基因(gal)和3-磷酸甘油脱氢酶基因(gpd)启动子分别为半乳糖、渗透压诱导型,在K. lactis中两者均存在较强的组成型表达,pScgal7在YEPG中活性提高57%,因此可通过添加诱导物调控外源基因的高效表达。根据已有报道[16],毕赤酵母中随着渗透压的提高,pScgpd1启动外源基因转录的能力有较大提升,但是其表达量仍较低。本实验中首次利用pScgpd11调控K. lactis外源蛋白的表达,在YEPD培养基中EGFP的表达量达到一个较高的水平,在高渗透压条件下其表达强度有所增加,表明渗透压对该启动子有一定的激活作用,在一定范围内可通过改变渗透压来调节外源蛋白的表达量,这有待进一步研究。pKlmal22起始转录受麦芽糖的诱导,Leifso等[17]研究发现,在葡萄糖存在的条件下,麦芽糖操纵子受到阻遏,本研究中,在YEPM中荧光强度高于YEPDM,可以间接证实这一点,但与上述几个启动子相比,该启动子表达水平较弱,可用于调控分泌过程中辅助因子、分子伴侣等的共表达。S. passalidarum是近年来分离得到的新型酵母,属于CTG类酵母[26, 27],与K. lactis亲缘关系较远,遗传系统的特殊性使得所选用启动子在K. lactis中调控外源蛋白表达时难以有效适用,启动活性受到影响,通过对该酵母启动元件进行分析有助于挖掘多样性启动子,但基于pSpmal1、pSpmal6、pSpgal1调控EGFP的表达处在较低水平,后续研究可以通过定点突变等基因改造方法研究S. passalidarum中性能优越的启动子在K. lactis中的调控表达。本研究在同一水平上比较不同启动子的功能,该评价体系具有一定的优势:(1) 以载体pKLAC1*为基础,保留了商业化质粒上lac4整合位点及amdS筛选标记,稳定整合于基因组中,通过替换不同启动子即可实现调控外源蛋白的表达;(2) 通过特异性引物PCR扩增方法筛选单拷贝整合重组菌,在同一基因剂量水平比较不同启动子活性差异;(3) 以egfp为报告基因,通过荧光显微镜和荧光分光光度计实现荧光强度定性和定量分析,快速直观。
基于上述不同启动子功能的分析及比较,下一步可以筛选启动强度高、易于进行调控、适用性广的启动子,同时可以针对特定启动子进行核心元件分析及上游增强子的分析,对启动子改造提供理论支撑,为今后在乳酸克鲁维酵母中高效表达外源蛋白奠定基础。
4 结论本研究以egfp为报告基因,比较pKladh4、pKlmal22、pScgal7、pScgpd1、pSpmal1、pSpmal6、pSpgal在K. lactis中的功能,并通过筛选单拷贝整合重组菌定性和定量分析胞内EGFP的表达,同时,在不同培养条件下分析了各启动子强度的变化。结果表明,pKladh4、pScgal7、pScgpd1具有较强起始外源基因转录的能力,并在相应诱导物存在条件下活性提高,均高于目前广泛应用的pKllac4启动子,为高效表达并调控外源蛋白提供更多的选择。
| [1] | Idiris A, Tohda H, Kumagai H, et al. Engineering of protein secretion in yeast:strategies and impact on protein production[J]. Applied Microbiology and Biotechnology, 2010, 86 (2): 403–417. DOI:10.1007/s00253-010-2447-0 |
| [2] | Swinkels BW, van Ooyen AJJ, Bonekamp FJ. The yeast Kluyveromyces lactis as an efficient host for heterologous gene expression[J]. Antonie van Leeuwenhoek, 1993, 64 (2): 187–201. DOI:10.1007/BF00873027 |
| [3] | Gellissen G, Hollenberg CP. Application of yeasts in gene expression studies:a comparison of Saccharomyces cerevisiae, Hansenula polymorpha and Kluyveromyces lactis-a review[J]. Gene, 1997, 190 (1): 87–97. DOI:10.1016/S0378-1119(97)00020-6 |
| [4] | Van Ooyen AJJ, Dekker P, Huang M, et al. Heterologous protein production in the yeast Kluyveromyces lactis[J]. FEMS Yeast Research, 2006, 6 (3): 381–392. DOI:10.1111/fyr.2006.6.issue-3 |
| [5] | Dujon B, Sherman D, Fischer G, et al. Genome evolution in yeasts[J]. Nature, 2004, 430 (6995): 35–44. DOI:10.1038/nature02579 |
| [6] | Spohner SC, Schaum V, Quitmann H, et al. Kluyveromyces lactis:An emerging tool in biotechnology[J]. Journal of Biotechnology, 2016, 222 : 104–116. DOI:10.1016/j.jbiotec.2016.02.023 |
| [7] | Heintzman ND, Ren B. The gateway to transcription:identifying, characterizing and understanding promoters in the eukaryotic genome[J]. Cellular and Molecular Life Sciences, 2007, 64 (4): 386–400. DOI:10.1007/s00018-006-6295-0 |
| [8] | Juven-Gershon T, Kadonaga JT. Regulation of gene expression via the core promoter and the basal transcriptional machinery[J]. Developmental Biology, 2010, 339 (2): 225–229. DOI:10.1016/j.ydbio.2009.08.009 |
| [9] | Alper H, Fischer C, Nevoigt E, et al. Tuning genetic control through promoter engineering[J]. Proceedings of the National Academy of Sciences of the United States of America, 2005, 102 (36): 12678–12683. DOI:10.1073/pnas.0504604102 |
| [10] | Niedenthal RK, Riles L, Johnston M, et al. Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast[J]. Yeast, 1996, 12 (8): 773–786. DOI:10.1002/(ISSN)1097-0061 |
| [11] | 吴沛桥, 巴晓革, 胡海, 等. 绿色荧光蛋白GFP的研究进展及应用[J]. 生物医学工程研究, 2009, 28(1): 83–86. |
| [12] | Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein(GFP)[J]. Gene, 1996, 173 (1): 33–38. DOI:10.1016/0378-1119(95)00685-0 |
| [13] | Saliola M, Mazzoni C, Solimando N, et al. Use of the KlADH4 promoter for ethanol-dependent production of recombinant human serum albumin in Kluyveromyces lactis[J]. Applied and Environmental Microbiology, 1999, 65 (1): 53–60. |
| [14] | Mazzoni C, Saliola M, Falcone C. Ethanol-induced and glucose-insensitive alcohol dehydrogenase activity in the yeast Kluyveromyces lactis[J]. Molecular Microbiology, 1992, 6 (16): 2279–2286. DOI:10.1111/mmi.1992.6.issue-16 |
| [15] | Maggi RG, Govind NS. Regulated expression of green fluorescent protein in Debaryomyces hansenii[J]. Journal of Industrial Microbiology and Biotechnology, 2004, 31 (7): 301–310. DOI:10.1007/s10295-004-0150-9 |
| [16] | 蒋慧慧, 李丰功, 陆毅, 等. 三种酵母启动子在毕赤酵母中的功能比较[J]. 中国生物工程杂志, 2011, 31(5): 60–68. |
| [17] | Leifso KR, Williams D, Hintz W E. Heterologous expression of cyan and yellow fluorescent proteins from the Kluyveromyces lactis KlMAL21-KlMAL22 bi-directional promoter[J]. Biotechnology Letters, 2007, 29 (8): 1233–1241. DOI:10.1007/s10529-007-9381-y |
| [18] | Bergkamp RJM, Kool IM, Geerse RH, et al. Multiple-copy integra-tion of the α-galactosidase gene from Cyamopsis tetragonoloba into the ribosomal DNA of Kluyveromyces lactis[J]. Current Genetics, 1992, 21 (4-5): 365–370. DOI:10.1007/BF00351696 |
| [19] | Madhavan A, Sukumaran RK. Promoter and signal sequence from filamentous fungus can drive recombinant protein production in the yeast Kluyveromyces lactis[J]. Bioresource Technology, 2014, 165 : 302–308. DOI:10.1016/j.biortech.2014.03.002 |
| [20] | Read JD, Colussi PA, Ganatra MB, et al. Acetamide selection of Kluyveromyces lactis cells transformed with an integrative vector leads to high-frequency formation of multicopy strains[J]. Applied and Environmental Microbiology, 2007, 73 (16): 5088–5096. DOI:10.1128/AEM.02253-06 |
| [21] | 刘波, 马清钧, 吴军. 乳酸克鲁维酵母表达外源蛋白研究进展[J]. 生物技术通讯, 2007, 18(6): 1039–1042. |
| [22] | 许强, 张梁, 李由然, 等. 腺苷酸脱氨酶在乳酸克鲁维酵母中构建表达[J]. 微生物学通报, 2016, 43(11): 2341–2352. |
| [23] | 唐瑞琪, 熊亮, 白凤武, 等. 酿酒酵母人工杂合启动子与天然启动子活性比较[J]. 生物技术通报, 2017, 33(1): 120–128. |
| [24] | Tanaka R, Ishibashi M, Tokunaga H, et al. Secretion of hen egg white lysozyme from Kluyveromyces lactis[J]. Bioscience, Biotechnology, and Biochemistry, 2000, 64 (12): 2716–2718. DOI:10.1271/bbb.64.2716 |
| [25] | Fermiñán E, Domínguez A. Heterologous protein secretion directed by a repressible acid phosphatase system of Kluyveromyces lactis:characterization of upstream region-activating sequences in the KIPHO5 Gene[J]. Applied and Environmental Microbiology, 1998, 64 (7): 2403–2408. |
| [26] | Laplaza JM, Torres BR, Jin YS, et al. Sh ble and Cre adapted for functional genomics and metabolic engineering of Pichia stipitis[J]. Enzyme and Microbial Technology, 2006, 38 (6): 741–747. DOI:10.1016/j.enzmictec.2005.07.024 |
| [27] | 范贺超. 新型木糖利用酵母的评价及其遗传表达系统构建[D]. 无锡: 江南大学, 2015. |