




2. 华智水稻生物技术有限公司,长沙 410125;
3. 湖南农业大学生物科学技术学院,长沙 410128
2. Huazhi Rice Biotech Co., Ltd., Changsha 410125;
3. College of Bioscience and Biotechnology, Hunan Agricultural University, Changsha 410128
对野生动植物的驯化和利用极大地推动了人类社会文明的进步,对植物种质资源遗传背景和多样性的认识与利用加速了植物育种技术的革命,从而为人类提供更多更优质的粮食和其他植物类产品。随着人口的增加、环境的恶化以及社会的发展,未来的粮食需求对包括水稻、麦类和玉米等主要粮食作物的育种提出了更高的要求,不仅要求稳产、高产,还要优质。在这种背景下,对优良种质资源的挖掘、培育和高效利用就成为现代育种的重要目标。因此,了解种质资源的进化、遗传背景和亲缘关系就成为杂种优势利用和品种改良的重要基础[1, 2]。
早期育种过程中对亲本遗传背景和亲缘关系的分析主要基于对其系谱和性状的考察,通过取长补短,优势互补,在杂交后代中筛选符合预期的表型性状,经过连续多代的选择以获得稳定的纯合系。我国育种学家开展了大量基于资料归纳和亲本调查的系谱分析工作[3-4],对促进我国主要作物的育种工作发挥了重要作用。
随着分子生物学技术的快速发展,对主要作物基因组的认识也日益全面和深入,人们开发了包括限制性片段长度多态性(Restriction fragment length polymorphism,RFLP)、多态性DNA随机扩增(Ra-ndom amplified polymorphic DNA,RAPD)、扩增片段长度多态性(Amplified fragment length polymorphism,AFLP)、微卫星DNA(Simple sequence repeats,SS-R)和单核苷酸多态性(Single nucleotide polymorph-ism,SNP)等在内的多种DNA水平的分子标记,并由此建立了多种基因型分型的方法,在指纹图谱构建、遗传多样性分析、种质资源鉴定和新等位基因的挖掘等方面得到了广泛应用[5, 6]。尤其是下一代高通量测序(Next generation sequencing,NGS)[7]和SNP分型技术[8]的发展,极大促进了对种质资源和重要性状相关等位基因的挖掘与鉴定,对提高育种过程中亲本选择与利用以及目标表型的筛选效率具有重要意义。
1 基因型分型的主要方法 1.1 传统的基因型分型方法传统的基因型分型方法主要基于早期开发的DNA水平的分子标记,如第一代以Southern杂交为基础发展起来的RFLP分子标记和第二代以PCR为基础发展起来的RAPD、AFLP和SSR等分子标记[9]。由于这些DNA分子标记具有与特定农艺和经济性状紧密连锁的特点,已被广泛应用于挖掘和克隆与特定农艺和经济性状相关的位点和基因组区段,使之在目标基因定位、标记辅助育种和基于图位克隆策略克隆新基因等研究中发挥着重要作用[10]。在这些分子标记中,SSR标记最早被用于植物育种。并且,随着分子生物学的发展和分子标记的开发利用,SSR等分子标记也被广泛应用于亲缘关系、遗传距离和遗传多样性分析等。但由于第一代RFLP分子标记的检测成本高和实验操作繁琐等特点而影响了其在实际使用中的推广。第二代分子标记中的RAPD标记技术的可靠性较差,AFLP标记则由于其技术的专利保护及操作程序长、步骤多、并要求很高的实验技能和精密的仪器设备等缺点,而难以被普及使用[9]。而且由于第一代和包括SSR等在内的第二代分子标记大多是在非编码区扩增或是在基因组中随机扩增,因此获得的多态性位点通常与目标性状基因的遗传距离较远,这就限制了这些标记在重要农艺性状基因的定位、图位克隆以及分子标记辅助育种中的应用[11]。随着高通量测序技术的广泛应用[7],推动了标记技术的发展,使得SNP等第三代分子标记逐渐居于主流地位[8]。
1.2 高通量的基因型分型方法近年来,随着群体遗传学和NGS技术的发展,越来越多的植物品种和品系的基因组重测序使SNP标记日益丰富,以其共显性、数量多、分布广和易于实现自动化分析等特点得到了日益广泛的研究和应用。为了进一步提高已有SNP的使用效率和分析通量,人们建立了竞争性等位基因特异性PCR[12](Competitive allele-specific PCR,KASP)技术和SNP芯片技术平台[13],促进了以SNP为核心的高通量基因型分型技术的发展,从而极大地推动了水稻、玉米、小麦等作物在遗传、种质鉴定和功能基因的克隆与鉴定、分子育种等方面的研究。
1.2.1 高通量测序技术利用NGS对作物种质资源的广泛重测序所获得的基因组数据,有助于深入挖掘SNP标记,从而全面分析不同品种之间的遗传差异,为在全基因组水平了解物种的进化、亲缘关系以及挖掘功能相关标记和基因提供了重要工具。深圳华大基因研究院较早开展了基于水稻高通量测序数据的SNP分析工作[14]。Yamamoto等[15]对水稻日本晴的近缘种越光进行了全基因组测序,并利用1 917个SNP位点对151个代表性的日本水稻材料进行了基因型分析。结果表明,60.9%的越光基因组具有原始祖先的单倍型,还发现现代育种技术降低了水稻的遗传多样性。Xu等[16]利用对40个栽培稻和10个野生稻的重测序数据,获得了650万个高质量的SNP位点,并在栽培稻中鉴定了大量低多样性的基因,可能位于驯化过程中受到人工选择的候选区域。
1.2.2 基于竞争性等位基因特异性PCR(KASP)的高通量SNP分型荧光染料EvaGreen等因为能以饱和的方式结合到双链DNA分子上,而且价格便宜,从而被广泛应用于实时定量PCR(Quantitative real-time PCR,qRT-PCR)[17]。随后,利用该荧光报告系统开发出了可以高通量检测SNP和InDel(Insertion-deletion)分子标记的KASP技术[12]。其原理是利用两条3’-末端碱基不同的等位基因正向引物分别对应检测位点的不同碱基状态,带有不同荧光的两条检测引物可以分别检测两条正向引物5’端的互补序列,经过与同一条反向引物PCR扩增以后,待测位点的多态性即可通过不同的荧光信号得以检测出来。该技术适用于高通量分析平台,可同时对大批量的SNP标记和样品进行精准的双等位基因验证和检测[18-19]。由于其通量高、稳定性好和价格便宜等优点,该技术已广泛应用于人、动物和植物遗传学研究与群体分型[20]。目前,英国LGC公司开发了最先进的基于KASP技术的SNPline分析平台。国内已有包括中国农业科学院、中国农业大学、中种集团和华智水稻生物技术有限公司等多家单位引进了这套系统。
1.2.3 基于SNP芯片的分型技术由于NGS的发展和成本的下降,更多物种资源被测序和重测序,这为高质量全基因组水平SNP的挖掘提供了便利,使得开发SNP芯片用于SNP的高通量检测成为一种趋势[13],并获得快速发展(表 1)。SNP芯片的工艺原理是在玻璃基片上通过光蚀刻的方式形成微米级的小孔,用以容纳微珠,每个微珠偶联上几十万个具有相同序列的DNA片段,该DNA片段游离的3’-端约50 bp的序列可与特定的SNP或InDel位点结合,芯片经过与待测样品的基因组DNA杂交以后检测杂交信号,即可在全基因组水平快速分析材料的遗传背景[21]。较早的全基因组SNP芯片开发是在樱桃上进行的[22]。随后,多个针对苹果、小麦和玉米等物种的芯片系统被陆续开发并广泛应用[23-26]。
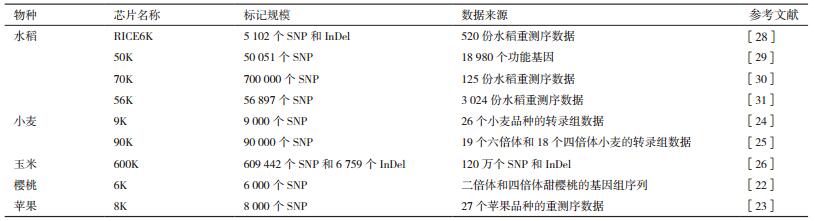
在水稻高通量检测和分型方面,邓兴旺[27]课题组曾指出基于组学如NGS和芯片技术的基因型分型平台的发展和应用将在分子设计育种实践中发挥重要作用。随后,人们又开发了多款水稻全基因组SNP芯片[28-30]。最近,华智水稻生物技术有限公司联合中国农业科学院开发了一款整合了56 897个SNP标记的56 K SNP芯片(未发表的数据),该芯片基于全球3 024份水稻样本的重测序数据[31]。
1.3 SNP分析工具和数据库随着越来越多作物品种被测序,SNP标记也日益丰富,为方便SNP数据的高效管理和有效利用,人们开始SNP分析工具和数据库的开发与研究(表 2)。较早期的AutoSNPdb数据库主要基于EST数据而开发,它包含了水稻、大麦和花椰菜等的SNP数据[32]。随后,一批通用性好、具有交互功能的SNP分析工具陆续得到了开发和应用[33-35]。近年来比较重要的水稻SNP分析平台主要有HapRice[36]、RiceVarMap[37]和IC4R[38]。HapRice主要提供SNP信息和检索功能。RiceVarMap提供基于标记名称、基因组区域、基因名称和基因注释等方式的标记查询。IC4R整合了来源于多个平台的水稻表达谱数据、同源检索数据库、水稻遗传多样性数据库和水稻相关文献挖掘等。CerealsDB是一款针对小麦(Triticum aestivum)SNP标记鉴定和分型的数据库[39]。

水稻作为我国最重要的粮食作物,对保障国家粮食安全具有不可替代的重要性。在新形势下,保障水稻高产、稳产和优质就成为水稻育种刻不容缓的目标。为此,近年来,人们利用NGS和水稻全基因组SNP芯片等高通量基因型分型技术结合全基因组关联分析(Genome-wide association study,GWAS),在水稻种质资源进化、重要农艺性状、抗病和耐逆相关位点的挖掘与鉴定方面取得了长足进展,极大促进了水稻功能基因组学的发展,为水稻育种提供了重要的参考信息。
2.1 水稻进化与人工选择研究在水稻人工选择和驯化方面,He等[40]对来自籼稻、粳稻和野生稻的66个水稻材料进行了重测序。结果显示,籼稻和粳稻有独立的起源祖先,且许多低多样性区域(LDRs)仅仅包含有一个已知的驯化基因,并鉴定出13个新的驯化相关基因。Lestari等[41]克隆了43个水稻品种的254条编码蔗糖合成酶3(RSUS3)的序列,通过SNP和InDel标记的分析最终鉴定了11个单倍型,发现了至少11个重组事件,且主要发生在转录区,为今后的遗传相关性研究建立了一种新的分类学方法。Yonemaru等[42]对177个日本水稻品种的遗传背景分析显示,由于育种选择导致在多个染色体区段的单倍型多样性下降,同时还发现由于人工选择而出现了新的单倍型多样性。这为通过基因聚合和人工选择来形成新的单倍型提供了理论基础。人们通常认为,现代野生稻是野生稻祖先群体的后代,野生稻的祖先群体衍生出现代驯化水稻。基于对203个驯化水稻和435个亚洲野生稻重测序数据的最新研究结果,Wang等[43]发现现代野生稻是一个杂合群体,这与野生稻和驯化水稻之间持续而广泛的通过花粉或种子介导的基因漂移有关。
2.2 水稻重要农艺性状相关位点分析近年来,利用GWAS策略挖掘水稻重要农艺性状的相关位点取得了重要进展。基于533份地方品种和杂交水稻的SNP标记,Yang等[44]进行了15个性状的GWAS分析,鉴定了一批与谷粒紧实度、谷粒投射面积等性状相关的SNP位点。对来自73个国家和地区的1 479个水稻品种的GWAS分析发现,大量与株高、叶夹角、开花时间、每株分蘖数、每穗粒数、粒重等性状相关的基因和位点在育种过程中受到了强烈选择[45]。Biscarini等[46]利用57 000个SNP标记对391个温带粳稻品种进行了GWAS分析,最终检测到42个基因型与表型之间的显著关联,其中控制剑叶宽度和株高的最显著关联分别位于4号和6号染色体。这些强关联的发现将可能应用于水稻的人工选择。最近,中科院植物生理生态研究所韩斌[47]课题组在杂种优势机理研究方面取得了重要进展。他们对17个代表性杂交水稻的F2代群体进行了重测序,获得了170万个SNP。随后的GWAS分析显示,尽管没有找到在所有材料中都存在的杂种优势相关位点,但在每个组内,有一些来自母本的基因组位点具有比父本更强的优势,同时还发现了产量相关的杂合位点和超亲杂种优势具有部分显性特征的证据。水稻早期生长势与积累、储藏和利用非结构性糖从而产生更大或更多叶片的基因型有关。Rebolledo等[48]利用12 221个SNP对123个日本水稻品种的早期生长势进行了GWAS分析,发现了9个与株高、营养器官的细胞大小或数目等相关的关联。
2.3 水稻耐逆和抗病相关研究挖掘对胁迫耐受的水稻相关基因和资源是目前水稻功能基因组学研究和育种的重要任务。Kumar等[49]利用SNP芯片对220个水稻品种的12个性状进行了GWAS分析,最终鉴定了20个与Na+/K+比值高度相关的SNP位点。Rahman等[50]利用376个SNP标记从107份水稻中最终鉴定出一批与已知耐盐品种不同的耐盐水稻资源。水稻对稻瘟病(Magnaporthe oryzae)的抗性是由许多主效基因或数量性状位点(Quantitative trait locus,QTL)来决定的。为了在全基因组水平探究水稻抗稻瘟病的分子机制,Kang等[51]利用水稻44K全基因组SNP芯片对420份水稻品种进行了稻瘟病抗性的GWAS分析,并利用含7万个SNP标记的芯片进行了验证,最终鉴定了97个稻瘟病抗性相关位点(LABRs)。其中82个为新位点,15个位点与已知抗性位点在同一区域。在LABRs区域的所有候选基因中,LABR_64与水稻对5个稻瘟病菌的抗性显著相关。
3 结语通过系谱法了解作物品种的遗传基础,追溯亲本来源,对于缩短作物育种进程,提高育种效率,充分利用杂种优势,具有重要意义。随着分子生物学的发展和基因组学与高通量分子标记技术的兴起,人们对作物遗传背景的分析有了更便捷的工具,如基于重测序数据的基因型分型方法(Genotyping-based Squencing,GBS)[7]和基于KASP[18-19]和SNP芯片[13]的高通量SNP分析技术,对育种群体目标性状的筛选和鉴定也有了更高效而科学的手段,而不再只依赖人工的田间性状观察和分析。但是重测序、KASP和SNP芯片的测试和数据分析成本比较高,而且对仪器设备和实验员的专业技术水平均有较高的要求,因此,此类实验需要有较大的项目支持,而且一般都由专业公司来完成。目前,国内已有多家公司和单位可以承担重测序和SNP高通量测试和分析任务,如华大科技的Illumina SNP芯片分型平台和Affymetrix基因分型平台,华智和中玉金标记的高通量分子育种平台,北京百迈克和上海欧易的全基因组重测序分析平台等。这些高通量分型技术的应用和专业技术平台的建立极大地推动了国内分子标记辅助选择[52]和基因组选择(Genomic selection,GS)[53]在动植物育种方面的应用,提升了种质资源多样性的利用水平,提高了育种效率。
同时,由于作物很多重要的经济和农艺性状往往受多基因或多位点影响,因此通过单个或者多个SNP对性状进行研究并不能取得理想的效果。而在进化和选择过程中有可能出现由多个功能相关SNP紧密连锁形成的单倍型,由于单倍型含有更多的连锁不平衡(Linkage disequilibrium,LD)信息可以挖掘,更有利于在关联分析(GWAS)中寻找和定位性状相关基因位点。利用GBS和高通量SNP分析技术,可以在全基因组水平获得自然群体进化、遗传多样性和单倍型等信息。因此,在全基因组水平上的基因型分型和单倍型分析与挖掘对基因聚合和作物品种改良具有重要的指导意义[42]。
| [1] |
Buckler ES, Gaut BS, McMullen MD. Molecular and functional diversity of maize[J]. Curr Opin Plant Biol, 2006, 9(2): 172-176. DOI:10.1016/j.pbi.2006.01.013 |
| [2] |
Collard BC, Mackill DJ. Marker-assisted selection:an approach for precision plant breeding in the twenty-first century[J]. Philos Trans R Soc Lond B Biol Sci, 2008, 363(1491): 557-572. DOI:10.1098/rstb.2007.2170 |
| [3] |
盖钧镒, 赵团结. 中国大豆育种的核心祖先亲本分析[J]. 南京农业大学学报, 2001, 24(2): 20-23. |
| [4] |
孙琦, 李文才, 于彦丽, 等. 美国商业玉米种质来源及系谱分析[J]. 玉米科学, 2016, 24(1): 8-13. |
| [5] |
McCouch S. Diversifying selection in plant breeding[J]. PLoS Biol, 2004, 2(10): e347. DOI:10.1371/journal.pbio.0020347 |
| [6] |
Slade AJ, Fuerstenberg SI, Loeffler D, et al. A reverse genetic, nontransgenic approach to wheat crop improvement by TILLING[J]. Nat Biotechnol, 2005, 23(1): 75-81. DOI:10.1038/nbt1043 |
| [7] |
Uitdewilligen J, Wolters AMA, D'hoop BB, et al. A next-generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato[J]. PLoS One, 2013, 8(5): e62355. DOI:10.1371/journal.pone.0062355 |
| [8] |
Munoz-Amatriain M, Cuesta-Marcos A, Hayes PM, et al. Barley genetic variation:implications for crop improvement[J]. Briefings in Functional Genomics, 2014, 13(4): 341-350. DOI:10.1093/bfgp/elu006 |
| [9] |
匡猛, 杨伟华, 许红霞, 等. 分子标记技术在棉花品种鉴定上的研究进展[J]. 棉花学报, 2009, 21(4): 330-334. |
| [10] |
Hayashi K, Hashimoto N, Daigen M, et al. Development of PCR-based SNP markers for rice blast resistance genes at the Piz locus[J]. Theor Appl Genet, 2004, 108(7): 1212-1220. DOI:10.1007/s00122-003-1553-0 |
| [11] |
王昊龙, 韩俊杰, 李淼淼, 等. 功能标记的开发及在禾谷类作物中的应用[J]. 核农学报, 2014, 28(11): 1963-1971. |
| [12] |
He C, Holme J, Anthony J. SNP genotyping:the KASP assay[J]. Methods Mol Biol, 2014, 1145: 75-86. DOI:10.1007/978-1-4939-0446-4 |
| [13] |
Ganal MW, Durstewitz G, Polley A, et al. A large maize(Zea mays L.)SNP genotyping array:development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome[J]. PLoS One, 2011, 6(12): e28334. DOI:10.1371/journal.pone.0028334 |
| [14] |
Li R, Li Y, Fang X, et al. SNP detection for massively parallel whole-genome resequencing[J]. Genome Res, 2009, 19(6): 1124-1132. DOI:10.1101/gr.088013.108 |
| [15] |
Yamamoto T, Nagasaki H, Yonemaru J, et al. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms[J]. BMC Genomics, 2010, 11: 267. DOI:10.1186/1471-2164-11-267 |
| [16] |
Xu X, Liu X, Ge S, et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes[J]. Nat Biotechnol, 2012, 30(1): 105-111. |
| [17] |
Li YD, Chu ZZ, Liu XG, et al. A cost-effective high-resolution melting approach using the EvaGreen dye for DNA polymorphism detection and genotyping in plants[J]. J Integr Plant Biol, 2010, 52(12): 1036-1042. DOI:10.1111/jipb.2010.52.issue-12 |
| [18] |
He C, Holme J, Anthony J. SNP genotyping:the KASP assay[J]. Methods Mol Biol, 2014, 1145: 75-86. DOI:10.1007/978-1-4939-0446-4 |
| [19] |
Zhou G, Zhang Q, Tan C, et al. Development of genome-wide InDel markers and their integration with SSR, DArT and SNP markers in single barley map[J]. BMC Genomics, 2015, 16(1): 804. DOI:10.1186/s12864-015-2027-x |
| [20] |
Graves H, Rayburn AL, Gonzalez-Hernandez JL, et al. Validating DNA polymorphisms using KASP assay in prairie cordgrass(Spartina pectinata Link)Populations in the U. S[J]. Front Plant Sci, 2015, 6: 1271. |
| [21] |
张小燕, 左明雪, 张占军, 等. 用基因芯片检测单核苷酸多态性反应原理[J]. 中国生物工程杂志, 2005, 25(11): 52-56. |
| [22] |
Peace C, Bassil N, Main D, et al. Development and evaluation of a genome-wide 6K SNP array for diploid sweet cherry and tetraploid sour cherry[J]. PLoS One, 2012, 7(12): e48305. DOI:10.1371/journal.pone.0048305 |
| [23] |
Chagne D, Crowhurst RN, Troggio M, et al. Genome-wide SNP detection, validation, and development of an 8K SNP array for apple[J]. PLoS One, 2012, 7(2): e31745. DOI:10.1371/journal.pone.0031745 |
| [24] |
Cavanagh CR, Chao S, Wang S, et al. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars[J]. PNAS, 2013, 110(20): 8057-8062. DOI:10.1073/pnas.1217133110 |
| [25] |
Wang S, Wong D, Forrest K, et al. Characterization of polyploid wheat genomic diversity using a high-density 90, 000 single nucleotide polymorphism array[J]. Plant Biotechnol J, 2014, 12(6): 787-796. DOI:10.1111/pbi.2014.12.issue-6 |
| [26] |
Unterseer S, Bauer E, Haberer G, et al. A powerful tool for genome analysis in maize:development and evaluation of the high density 600 k SNP genotyping array[J]. BMC Genomics, 2014, 15: 823. DOI:10.1186/1471-2164-15-823 |
| [27] |
Chen H, He H, Zhou F, et al. Development of genomics-based genotyping platforms and their applications in rice breeding[J]. Curr Opin Plant Biol, 2013, 16(2): 247-254. DOI:10.1016/j.pbi.2013.04.002 |
| [28] |
Yu H, Xie W, Li J, et al. A whole-genome SNP array(RICE6K)for genomic breeding in rice[J]. Plant Biotechnol J, 2014, 12(1): 28-37. DOI:10.1111/pbi.12113 |
| [29] |
Singh N, Jayaswal PK, Panda K, et al. Single-copy gene based 50 K SNP chip for genetic studies and molecular breeding in rice[J]. Sci Rep, 2015, 5: 11600. DOI:10.1038/srep11600 |
| [30] |
McCouch SR, Wright MH, Tung CW, et al. Open access resources for genome-wide association mapping in rice[J]. Nat Commun, 2016, 7: 10532. DOI:10.1038/ncomms10532 |
| [31] |
Li JY, Wang J and Zeigler RS. The 3, 000 rice genomes project:new opportunities and challenges for future rice research[J]. Gigascience, 2014, 3: 8. DOI:10.1186/2047-217X-3-8 |
| [32] |
Duran C, Appleby N, Clark T, et al. AutoSNPdb:an annotated single nucleotide polymorphism database for crop plants[J]. Nucleic Acids Res, 2009, 37(suppl_1): D951-953. |
| [33] |
Nijveen H, van Kaauwen M, Esselink DG, et al. QualitySNPng:a user-friendly SNP detection and visualization tool[J]. Nucleic Acids Res, 2013, 41(W1): W587-590. DOI:10.1093/nar/gkt333 |
| [34] |
Azam S, Rathore A, Shah T M, Telluri M, et al. An integrated SNP mining and utilization(ISMU)pipeline for next generation sequencing data[J]. PLoS One, 2014, 9(7): e101754. DOI:10.1371/journal.pone.0101754 |
| [35] |
Dereeper A, Homa F, Andres G, et al. SNiPlay3:a web-based application for exploration and large scale analyses of genomic variations[J]. Nucleic Acids Res, 2015, 43(W1): W295-300. DOI:10.1093/nar/gkv351 |
| [36] |
Yonemaru J-i, Ebana K, Yano M. HapRice, an SNP Haplotype Database and a Web Tool for Rice[J]. Plant Cell Physiol, 2014, 55(1): e9. DOI:10.1093/pcp/pct188 |
| [37] |
Zhao H, Yao W, Ouyang Y, et al. RiceVarMap:a comprehensive database of rice genomic variations[J]. Nucleic Acids Res, 2015, 43(D1): D1018-1022. DOI:10.1093/nar/gku894 |
| [38] |
Consortium IRP. Information Commons for Rice(IC4R)[J]. Nucleic Acids Res, 2016, 44(D1): D1172-1180. DOI:10.1093/nar/gkv1141 |
| [39] |
Wilkinson PA, Winfield MO, Barker GL, et al. CerealsDB 3. 0:expansion of resources and data integration[J]. BMC Bioinformatics, 2016, 17: 256. DOI:10.1186/s12859-016-1139-x |
| [40] |
He Z, Zhai W, Wen H, et al. Two evolutionary histories in the genome of rice:the roles of domestication genes[J]. PLoS Genet, 2011, 7(6): e1002100. DOI:10.1371/journal.pgen.1002100 |
| [41] |
Lestari P, Lee G, Ham TH, et al. Single nucleotide polymorphisms and haplotype diversity in rice sucrose synthase 3[J]. J Hered, 2011, 102(6): 735-746. DOI:10.1093/jhered/esr094 |
| [42] |
Yonemaru J, Yamamoto T, Ebana K, et al. Genome-wide haplotype changes produced by artificial selection during modern rice breeding in Japan[J]. PLoS One, 2012, 7(3): e32982. DOI:10.1371/journal.pone.0032982 |
| [43] |
Wang H, Vieira FG, Crawford JE, et al. Asian wild rice is a hybrid swarm with extensive gene flow and feralization from domesticated rice[J]. Genome Res, 2017, 27: 1-10. DOI:10.1101/gr.202945.115 |
| [44] |
Yang W, Guo Z, Huang C, et al. 15 genetic variation in rice[J]. Nat Commun, 2014, 5: 5087. DOI:10.1038/ncomms6087 |
| [45] |
Xie W, Wang G, Yuan M, et al. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection[J]. Proc Natl Acad Sci USA, 2015, 112(39): E5411-5419. DOI:10.1073/pnas.1515919112 |
| [46] |
Biscarini F, Cozzi P, Casella L, et al. Genome-Wide Association Study for Traits Related to Plant and Grain Morphology, and Root Architecture in Temperate Rice Accessions[J]. PLoS One, 2016, 11(5): e0155425. DOI:10.1371/journal.pone.0155425 |
| [47] |
Huang X, Yang S, Gong J, et al. Genomic architecture of heterosis for yield traits in rice[J]. Nature, 2016, 537(7622): 629-633. DOI:10.1038/nature19760 |
| [48] |
Rebolledo M C, Dingkuhn M, Courtois B, et al. Phenotypic and genetic dissection of component traits for early vigour in rice using plant growth modelling, sugar content analyses and association mapping[J]. J Exp Bot, 2015, 66(18): 5555-5566. DOI:10.1093/jxb/erv258 |
| [49] |
Kumar V, Singh A, Mithra SV, et al. Genome-wide association mapping of salinity tolerance in rice(Oryza sativa)[J]. DNA Res, 2015, 22(2): 133-145. DOI:10.1093/dnares/dsu046 |
| [50] |
Rahman MA, Thomson MJ, Shah EAM, et al. Exploring novel genetic sources of salinity tolerance in rice through molecular and physiological characterization[J]. Ann Bot, 2016, 117(6): 1083-1097. DOI:10.1093/aob/mcw030 |
| [51] |
Kang H, Wang Y, Peng S, et al. Dissection of the genetic architecture of rice resistance to the blast fungus Magnaporthe oryzae[J]. Mol Plant Pathol, 2016, 17(6): 959-972. DOI:10.1111/mpp.12340 |
| [52] |
宁洽, 刘文国, 杨伟光, 等. SNP标记在玉米研究上的应用进展[J]. 玉米科学, 2017, 25(1): 57-61. |
| [53] |
吕锐玲, 周强. 植物基因组选择及其应用研究进展[J]. 中国农学通报, 2016, 32(15): 107-11. DOI:10.11924/j.issn.1000-6850.casb15110089 |