




2. 山东农业大学作物生物学国家重点实验室, 泰安271000
2. State Key Laboratory of Crop Biology, Shandong Agricultural University, Tai'an 271000
微生物是海洋生态系统中的重要组成部分,蕴藏着巨大的生物量,在碳、氮、硫及磷等元素的全球生物地球化学循环,净化海洋环境及维持海洋生态系统多样性与稳定性方面发挥着重要作用[1-3]。虽然海洋微生物在生态系统中具有如此重要的作用,但目前人们对其了解仍相对缺乏。对海洋微生物多样性和分布特征的研究将有助于更深入地认识海洋微生物在整个海洋生态系统中的功能与作用,对开展海洋生态环境研究具有重要意义。
早期微生物生态研究是通过传统纯培养获得菌株后才能对其特性进行描述[4]。但由于培养条件与自然条件的差异,实际培养所得到的只是自然环境中的少部分(0.001%-15%)微生物[5]。近年来,随着现代分子生物学技术的发展,使直接检测环境样品中的未培养微生物群落成为可能。这些技术包括:构建16S rDNA克隆文库、末端限制性片段长度多态性(T-RFLP)、变性梯度凝胶电泳(DGGE)、高通量测序技术[6]等。目前,应用最广泛的是克隆文库法和高通量测序技术,两者各有优劣,前者不受序列长度限制,缺陷是通量较低、花费较高;后者通量高、价格相对便宜,但测序长度受限[6]。
黄河是世界上含沙量最高的河流,平均含沙量为22 g/L[7]。黄河入海口,位于黄河与渤海湾交汇处,黄河携带的大量泥沙、污染物,以及丰富的营养物质在此汇入海洋。虽然前人对黄河入海口地区微生物多样性已有少量研究,但对该独特的入海口生态系统的研究还不充分。Li等[8]研究了黄河口表层水体中反硝化细菌功能基因的丰度及多样性分布。基于nirK基因的研究显示,淡水环境中反硝化细菌群落多样性高于海水环境,同时反硝化群落组成与多种环境因素有关。Yan等[9]研究了黄河入海口沉积物中亚硝酸盐依赖型厌氧甲烷氧化细菌(n-damo)的多样性。研究发现,黄河口n-damo细菌多样性相对高于其他生境。Wei等[10]对比研究了黄河入海口沉积物及水体中细菌和古菌群落的丰度、多样性及其分布模式。研究表明,黄河口沉积物和上覆水中细菌和古菌的分布模式不同。本研究旨在运用16S rDNA克隆文库技术探索黄河入海口不同生境水体细菌群落多样性及空间分布特征。研究结果将对该地区微生物资源的开发、黄河水资源利用及生态系统保护具有一定的理论和实际意义。
1 材料与方法 1.1 材料 1.1.1 采样点及样品采集2010年10月下旬,于黄河入海口不同地理位置进行样品采集,其中两采样点(A、B)位于正对河口的河海交汇处,另两样点(C、D)为离河口相对较远的近岸处(图 1)。用水样采集器(Wildco,美国)采集表层水样,装于无菌采样瓶中,水温、盐度、pH、溶解氧等理化指标,用便携式仪器(雷磁,上海)现场测定。

|
| 图 1 黄河入海口及采样点地理位置分布图 采样点A(E119°15'26.5'',N37°51'10.2'');B(E119°09'20.4'',N37°45'37.1'')为正对河口的河海交汇处;C(E119°18'20.4'',N37°43'54.8'')为离河口相对较远的海水养殖区;D(E119°18'29.0'',N37°43'49.5'')为海上油井旁海水 |
10×PCR Buffer、2.5 mmol/L dNTP、Taq DNA Polymerase(TaKaRa,大连);Hae Ⅲ、Msp Ⅰ限制性内切酶及pMD18-T载体(TaKaRa);大肠杆菌DH5α感受态细胞(天根,北京);DNA提取试剂盒E.N.Z.A.TMWater DNA Kit(Omega,美国);琼脂糖凝胶DNA回收试剂盒(Solarbio,北京);琼脂糖(BIOWEST,西班牙)。
1.2 方法 1.2.1 样品处理样品用冰盒低温运回实验室后,分别将各样点的部分水样,分装冻存(-20℃),并尽快参照国标方法对总氮、硝态氮、总磷、化学需氧量(COD)等理化指标进行测定。另一部分水样分别抽滤至孔径为0.22 μm的无菌滤膜上,保存于-80℃超低温冰箱,以备DNA提取。
1.2.2 16SrDNA克隆文库的构建 1.2.2.1 DNA提取及检测参照说明书,用E.N.Z.A.TM Water DNA Kit试剂盒分别对不同样点的DNA进行提取,所得DNA样品经1%(m/V)琼脂糖凝胶电泳对DNA质量进行检测。
1.2.2.2 16S rDNA目的片段扩增用细菌16S rDNA通用引物27F和1492R对各点DNA样品进行PCR扩增。PCR反应体系及条件参照Wei等[11]文献报道。PCR产物用1%的琼脂糖凝胶电泳检测,目的片段纯化用琼脂糖凝胶DNA回收试剂盒参照说明书进行。
1.2.2.3 16S rDNA文库构建将纯化后的PCR产物与pMD18-T载体连接并转化入大肠杆菌DH5α感受态细胞中。培养2 h后,取100 μL菌液涂布至含有氨苄青霉素的LB琼脂平板上,37℃培养过夜。随机挑取单克隆,在含有氨苄青霉素的LB液体培养基中培养至指数期,用载体引物(RV-M和M13-47) 通过菌液直接为模板的PCR筛选阳性克隆,1%琼脂糖凝胶电泳检测PCR结果。
1.2.2.4 RFLP分析及测序将阳性克隆PCR产物,参照说明书分别用限制性核酸内切酶Hae Ⅲ和Msp Ⅰ进行酶切。酶切结束后用3%的琼脂糖凝胶电泳检测酶切结果。两种酶切带型均一致的克隆被认为是同一物种。挑选酶切带型不同的阳性克隆,分别用载体引物进行双端测序,拼接得到16S rDNA近全长序列。
1.2.3 16SrDNA克隆文库数据分析对所得16S rDNA序列先去除载体序列、统一序列方向,剔除序列长度较短、引物序列缺失及存在模糊碱基的序列。用mothur(www.mothur.org)去除嵌合体序列,以97%序列相似性为阈值进行OTU(Operational Taxonomic Unit)划分。挑选OTU中丰度最高的序列为代表OTU序列,以SILVA数据库(www.arb-silva.de)为参考对代表序列进行物种分类,置信阈值为0.8。结合RFLP结果,整理各样点不同分类水平物种分类表。不同分类水平群落相似性聚类树及多样性指数计算用PAST软件(http://folk.uio.no/ohammer/past/)进行,分别用Shannon指数、Evenness指数和Chao 1指数来表示多样性、均匀度和丰富度估测。维恩图用在线工具Venny 2.1完成(http://bioinfogp.cnb.csic.es/tools/venny/index.html)。选取优势OTU代表序列(序列数相对丰度 > 1%)及其在NCBI及EZ BioCloud(http://www.ezbiocloud.net/)中的近缘序列用MEGA软件(http://www.megasoftware.net/)进行系统发育树构建。根据物种分类结果,用FAPROTAX数据库(http://www.zoology.ubc.ca/louca/FAPROTAX/lib/php/index.php?sectin=Home)在QⅡME中参照网站说明对细菌群落功能进行注释。环境因子与优势类群及群落功能间Spearman相关性分析用SPSS软件(IBM,美国)完成。环境因子与群落组成及功能轮廓间的冗余分析(RDA)用CANOCO软件(http://www.canoco5.com/)进行。
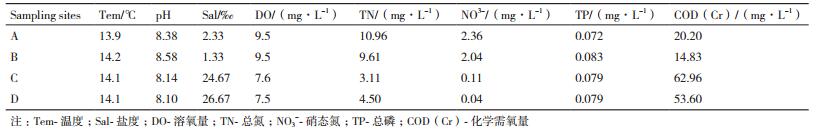
2 结果 2.1 采样点及样品理化指标由理化指标可知,正对河口处A、B两点,盐度较低、溶氧量较高;离河口相对较远处C、D两点,盐度较高,溶氧量较低。此外,A、B两点的pH、总氮及硝态氮含量均高于C、D两点,COD(化学需氧量)明显低于C、D两点,总磷含量相近(表 1)。因此,根据地理位置和理化指标,可将采样点生境明显分为受黄河水影响较大的河海交汇低盐区(A、B)和受河水影响较小的海水高盐区(C、D)两类。
针对不同的采样点共构建了4个不同的16S rDNA克隆文库,每个文库随机挑取了120个单菌落,其中阳性克隆数平均100个左右,阳性克隆率约85%。经过RFLP及克隆文库测序,共获得408条序列,按照97%序列相似性可划分为178个OTU。由表 2可知,各点覆盖率(Coverage)为0.60-0.68,表明所建文库能较准确地反映各样点的优势细菌类群,但尚有大量稀有种未被检测到。河海交汇处样点B的细菌均匀度(Evenness)、Chao 1及Shannon指数均最高,表明B点细菌多样性最高。海水养殖区C点的均匀度指数最低,表明其细菌群落组成不均匀,存在优势度较高的细菌类群。比较不同点的Shannon多样性指数,发现河海交汇处低盐区(A、B)细菌多样性整体高于高盐区(C、D)。
通过16S rDNA克隆文库法,在黄河入海口水体中共检测到分类地位明确的11个门、18个纲、39个科、53个属的细菌。通过门到属水平的细菌群落组成相似性聚类,发现黄河口水体细菌群落在各分类水平上均可聚为明显的两个分支——A、B和C、D(图 2)。

|
| 图 2 黄河入海口水体细菌群落结构分布图 A:(门水平)细菌群落结构分布图;B:(纲水平)细菌群落结构分布图;C:(科水平)细菌群落结构分布图;D:(属水平)细菌群落结构分布图 |
在门水平上,相对丰度 > 1%的优势门有变形菌门(Proteobacteria,56%-79%)、放线菌门(Actino-bacteria,8%-30%)、拟杆菌门(Bacteroidetes,4%-11%)、蓝细菌门(Cyanobacteria,1%-3%)、浮霉菌门(Planctomycetes,1%-3%)等11个。其中A、B样点放线菌门相对丰度明显高于C、D样点,但变形菌门及拟杆菌门的相对丰度则低于C、D样点(图 2-A)。纲水平上,优势纲有β-变形菌纲(Betaprote-obacteria,33%-43%)、α-变形菌纲(Alphaproteoba-cteria,14%-27%)、放线菌纲(Actinobacteria,5%-30%)、γ-变形菌纲(Gammaproteobacteria,4%-17%)及黄杆菌纲(Flavobacteria,1%-10%)等18个。其中A、B样点中放线菌纲的相对丰度高于C、D样点,α-变形菌纲和黄杆菌纲相对丰度则低于C、D样点(图 2-B)。科水平上,优势科为从毛单胞菌科(Comamonadaceae,14%-39%)、鱼孢菌科(Sporichthyaceae,1%-22%)、Pelagibacteraceae科(6%-16%)、酸微菌科(Acidimicrobiaceae,4%-16%)和红杆菌科(Rhodobacteraceae,2%-16%)等39个。其中A、B样点中鱼孢菌科和酸微菌科的相对丰度明显高于C、D样点,从毛单胞菌科和红杆菌科相对丰度则低于C、D样点(图 2-C)。在属水平上,优势属有BAL58 marine group(0%-31%)、hgcI clade(0%-21%)、CL500-29 marine group(4%-15%)、红育菌属(Rhodoferax,4%-11%)及LD28(0%-9%)等53个。A、B样点中除BAL58 marine group外,其余优势属的相对丰度均高于C、D样点(图 2-D)。
为进一步揭示OTU水平的细菌群落分布特征,绘制了OTU水平的韦恩图。结果(图 3)表明,不同样点以独有OTU为主,A、B、C、D各点独有OTU数目分别为39、35、38、43个。相似环境的共有OTU数目更多,同为河口处低盐区的A、B样点共有15个OTU,高盐区的C、D样点共有7个OTU。但A与C、A与D、B与C、B与D之间共有OTU数目分别仅为2、1、3、1个。有2个OTU在A、B、C样点中均有分布;仅有1个OTU在A、B、D样点中均有分布,未检测到OTU在所有样点中均存在。

|
| 图 3 黄河入海口细菌群落OTU水平分布维恩图 |
根据物种分类结果,对黄河入海口水体细菌群落用FAPROTAX数据库进行功能注释(图 4)。与细菌群落组成类似,从功能类群层面,黄河口细菌群落也可聚为明显的两个分支——A、B和C、D。黄河口水体最优势的功能类群为好氧化能异养菌(7%-23%)、其次为光能异养菌(0%-7%)、甲基营养型(0%-7%)、硝酸盐还原菌(0%-7%)等,硫元素、重金属、芳香烃类难降解有机物代谢及人或哺乳动物肠道菌群等也占有一定比例。其中A、B样点的光能异养和甲基营养型菌的比例高于C、D样点;而好氧化能异养和硝酸还原类群低于C、D样点。

|
| 图 4 黄河入海口水体细菌功能注释热图 颜色深浅表示功能类群所对应的序列相对丰度大小 |
挑选相对丰度 > 1%的优势OTU进行系统进化分析,以确定其分类地位、可培养状况及近缘序列环境来源(图 5)。通过与EZ BioCloud数据库比对,可知OTU1、2、3、4、5、9、12、13、14和18已有序列相似性 > 97%的可培养株,OTU6、7、8和15相似性最高的序列为来自环境的未培养细菌,表明即使是优势OTU水平,黄河口水体中仍有一定比例的未培养细菌新种。同时我们发现,OTU1、3、5、13和14的近缘序列菌株来源为海水环境,上述OTU全部来自C、D两个海水样点。OTU2、6、7、9、15与18的近缘序列菌株来源为淡水环境,它们大部分来自A、B两个低盐样点。其中A点所占比例最大的OTU2为α-变形菌纲的细菌,在NCBI中其相似度最高的序列来自巴拿马的加通湖表面温水层。B点所占比例最大的OTU6为一种不可培养的细菌,其相似度最高的序列也来自巴拿马的加通湖表面温水层。在C、D两点所占比例均最大的OTU1为β-变形菌纲的细菌,其相似度最高的序列来自美国纽波特港水环境,该地盐度与C、D两点相近。

|
| 图 5 根据优势OTU的16S rDNA序列构建的系统发育树 分支末端展示的为优势OTU编号及GenBank登录号;红色标注表示本研究中OTU,小括号中展示该OTU的分布样点及序列数;蓝色为可培养近缘序列,黑色为目前未培养近缘序列,中括号中依次展示了序列相似性值、分类地位及样品来源等信息。分支上的数字为自展值百分比且显示>80 %;线段0.2为核苷酸替换率 |
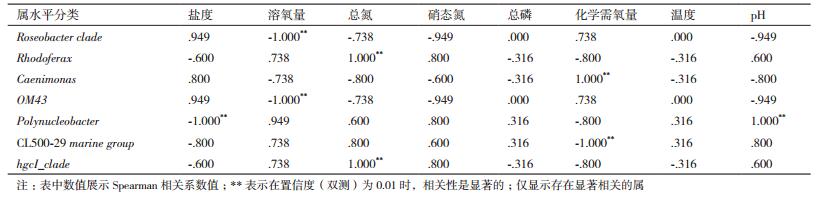
优势属及功能类群与理化指标间的斯皮尔曼相关性分析显示,仅有部分优势属(玫瑰杆菌属、红育菌属、Caenimonas属等)与盐度、溶氧量、总氮及COD等理化因子显著相关(表 3),而绝大部分优势功能类群(甲醇氧化型细菌、甲基营养菌、好氧光能异养菌等)与盐度、溶氧量、总氮、硝态氮等理化因子显著相关(表 4)。

分别以黄河入海口水体细菌属水平群落组成、群落功能组成和所检测的理化因子进行冗余分析(RDA),研究环境因子与整体细菌群落结构及功能之间的关系。结果(图 6)显示,属水平及功能角度分析结果一致,即A、B样点均有聚集现象,其群落及功能分布均与溶氧量及总氮正相关,与盐度负相关,而C、D样点的细菌群落及功能分布则与上述理化因子的关系正好相反。

|
| 图 6 黄河入海口水体细菌群落与理化因子冗余分析 A:黄河入海口水体细菌属水平群落与环境因子RDA排序图;B:黄河入海口水体细菌功能群落与环境因子RDA排序图 |
通过对黄河河口及其邻近海域水体的16S rDNA克隆文库的构建及多样性分析,我们对该地区细菌群落结构及功能轮廓有了更完善的了解。其中河海交汇处低盐区的水体细菌多样性相对高盐区海水的细菌多样性高。推测河海交汇处细菌多样性高的原因有两个:一是汇集作用,即沿水流方向,细菌的丰度及多样性增加[11]。该区域是淡水和海水的交汇处,淡水和海水来源的细菌在此汇集,因而使得河海交汇处细菌多样性较高。二是盐度等环境因子影响。前人研究表明,盐度是影响入海口水体细菌群落结构及多样性的关键因子[12-13]。Campbell等[13]通过对沿入海口盐度梯度的水体细菌群落研究,发现低盐度( < 5‰)和高盐度( > 30‰)水体细菌多样性明显高于中等盐度(5‰-30‰)水体,这与本文的研究结果一致。此外,前人研究还指出溶解氧、pH、盐度及与营养相关的变量(硝酸盐和磷酸盐)是驱动沿海水体细菌群落多样性的关键因素[14]。在本研究中,盐度、pH、溶解氧、硝酸盐和氨浓度也与黄河入海口浮游细菌群落密切相关。
本研究一个重要的发现是,无论从物种组成层面还是功能角度,样品均聚为差异明显的两个分支——A、B和C、D。正如前人研究所述,环境条件的差异对水环境中微生物群落的组成和功能产生影响[15-17]。本研究中,与采样点位置差异引起的显著理化差异密切相关。A、B两点正对河口,位于河海交汇处,河水的扰动使得溶解氧含量较高,稀释作用使得盐度较低,外加携带上游农田的肥料而氮含量较高;C为海上养殖区,饵料投放、水产排便等使得改点COD含量较高;D位于海上油井旁,来自采油过程中的污染使得该点COD含量也较高;C、D两点均离河口相对较远,因此受河水影响小,盐度高。上述因素,使得A、B和C、D生境差异明显,因而驱动了其微生物群落组成及功能轮廓的显著差异。系统进化分析从物种潜在来源的角度进一步阐明了不同位点的群落差异。A、B两点优势OTU相似性最高的序列均来自湖泊等淡水环境,因此推测A、B优势群落可能来自黄河水。C、D点优势OTU的相似序列均来自海水环境,表明优势类群主要来自海洋环境。
水体细菌对维持黄河入海口生态系统平衡至关重要。本研究中优势类群主要参与黄河口碳、氮、硫等元素循环代谢。在属水平上我们发现黄河口主要优势类群为hgcI clade、CL500-29 marine group和红育菌属。根据前人研究报道,CL500-29 marine group具有氨氧化及水解尿素的功能[18],红育菌属细菌具有铁还原作用[19]及固氮能力[20],这些功能在基于FAPROTAX的功能注释中均有体现。同时,功能注释结果显示,A、B、C、D点好氧化能异养菌丰度最高,尤其是C、D点,这可能与其COD含量高有关,高比例的化能异养菌有助于有机质的降解,从而有效降低COD。甲基营养型细菌在各点均有较高比例,这类细菌可能在黄河口水体中含甲基有机物代谢中发挥关键作用。有趣的是,C、D点海水样品中检测到高比例的硝酸盐还原类群但A、B低盐样点则未检测到,表明细菌反硝化作用可能在黄河口海水硝酸盐去除中发挥重要作用,低盐环境下水体硝酸盐去除可能主要通过其他途径。A、B点还注释到丰富的尿素水解类群(ureolysis),这与黄河水汇集农田氮肥引起该区域高浓度氮素相关。此外,A、B样点还注释到了人及哺乳动物肠道来源的菌群,C、D点注释到了丰富的硫代谢相关菌群,这都与采样点的特殊环境密切相关。此外,在分析环境因子与优势细菌类群的关系中发现,按照微生物功能划分的功能类群相比按照16S rDNA序列相似性划分的物种组成,对环境因子响应更敏感。这与前人在全球尺度海洋环境[21]、模拟草原土壤风蚀与沉积[22]、凤梨科植物储水器[23]等不同生境微生物群落与环境因子关系的研究中结果一致。河口、海洋、土壤、植物储水器等多样化生境下一致的结果表明,微生物功能类群相比物种分类对环境因子响应更敏感的结论可能具有广泛的普适性。
本文主要研究了黄河入海口水体细菌群落多样性及空间分布特征。为初步掌握黄河河口及其邻近海域水体细菌多样性状况及功能轮廓提供了一定的参考,阐明了影响该区域内细菌多样性分布的环境因子(溶解氧、pH、氮营养盐),对进一步改善该区域河流和海洋环境提供了数据支持。本研究的主要优势在于使用克隆文库法可得到16S rDNA的近全长序列,细菌分类注释结果更准确。但尚有不足,如克隆文库覆盖率低,不能获得稀有种信息,后续可结合高通量测序进行分析;样点相对较少,后续可开展针对该地区更为密集的采样研究;缺少时序性,进一步的研究中可增加不同季节的采样。此外,本研究还揭示了该地区尚存在大量未培养的细菌资源,后续还要结合新技术(如宏基因组/宏转录组测序)加强对该地区功能微生物类群及细菌资源的开发。
4 结论本研究通过对黄河入海口不同位点水体细菌群落结构及功能的研究,发现优势细菌类群主要与碳、氮、硫等元素循环代谢相关,且优势类群中仍然存在大量未可培养的细菌。水体理化性质差异是引起水体细菌群落结构及功能组成差异的主要驱动因素之一。细菌功能轮廓相比其群落结构,对环境因子的响应更为敏感。
| [1] | Mou X, Sun S, Edwards RA, et al. Bacterial carbon processing by generalist species in the coastal ocean[J]. Nature, 2008, 451 (7179): 708–711. DOI:10.1038/nature06513 |
| [2] | Glaubitz S, Lueders T, Abraham WR, et al. 13C-isotope analyses reveal that chemolithoautotrophic Gamma-and Epsilonproteobacteria feed a microbial food web in a pelagic redoxcline of the central Baltic Sea[J]. Environ Microbiol, 2009, 11 (2): 326–337. DOI:10.1111/emi.2009.11.issue-2 |
| [3] | Hargrave BT, Holmer M, Newcombe CP. Towards a classification of organic enrichment in marine sediments based on biogeochemical indicators[J]. Marine Pollution Bulletin, 2008, 56 (5): 810–824. DOI:10.1016/j.marpolbul.2008.02.006 |
| [4] | Anderson IC, Cairney J WG. Diversity and ecology of soil fungal communities:increased understanding through the application of molecular techniques[J]. Environ Microbiol, 2004, 6 (8): 769–779. DOI:10.1111/emi.2004.6.issue-8 |
| [5] | Schleifer KH. Microbial diversity:facts, problems and prospects[J]. Systematic and Mpplied Microbiology, 2004, 27 (1): 3–9. DOI:10.1078/0723-2020-00245 |
| [6] | 姬洪飞, 王颖. 分子生物学方法在环境微生物生态学中的应用研究进展[J]. 生态学报, 2016, 36(24): 8234–8243. |
| [7] | Xia N, Xia X, Liu T, et al. Characteristics of bacterial community in the water and surface sediment of the Yellow River, China, the largest turbid river in the world[J]. Journal of Soils and Sediments, 2014, 14 (11): 1894. DOI:10.1007/s11368-014-0974-5 |
| [8] | Li J, Wei G, Wang N, et al. Diversity and distribution of nirK-harboring denitrifying bacteria in the water column in the Yellow River estuary[J]. Microbes and Environments, 2014, 29 (1): 107–110. DOI:10.1264/jsme2.ME13111 |
| [9] | Yan P, Li M, Wei G, et al. Molecular fingerprint and dominant environmental factors of nitrite-dependent anaerobic methane-oxidizing bacteria in sediments from the Yellow River Estuary, China[J]. PLoS One, 2015, 10 (9): e0137996. DOI:10.1371/journal.pone.0137996 |
| [10] | Wei G, Li M, Li F, et al. Distinct distribution patterns of prokaryotes between sediment and water in the Yellow River estuary[J]. Applied Microbiology and Biotechnology, 2016, 100 (22): 9683–9697. DOI:10.1007/s00253-016-7802-3 |
| [11] | Wei G, Li J, Wang N, et al. Spatial abundance and diversity of Bacterioplankton in a typical stream-forming ecosystem, Huangqian Reservoir, China[J]. J Microbiol Biotechnol, 2014, 24 (10): 1308–1318. DOI:10.4014/jmb.1403.03067 |
| [12] | Kirchman DL, Cottrel MT, DiTullio GR. Shaping of bacterial community composition and diversity by phytoplankton and salinity in the Delaware Estuary, USA[J]. Aquatic Microbial Ecology, 2017, 78 (2): 93–106. DOI:10.3354/ame01805 |
| [13] | Campbell BJ, Kirchman DL. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient[J]. The ISME journal, 2013, 7 (1): 210. DOI:10.1038/ismej.2012.93 |
| [14] | Wang K, Ye X, Chen H, et al. Bacterial biogeography in the coastal waters of northern Zhejiang, East China Sea is highly controlled by spatially structured environmental gradients[J]. Environ Microbiol, 2015, 17 (10): 3898–3913. DOI:10.1111/1462-2920.12884 |
| [15] | Kim J, Kim HS, Han S, et al. Hydrodynamic effects on bacterial biofilm development in a microfluidic environment[J]. Lab on a Chip, 2013, 13 (10): 1846–1849. DOI:10.1039/c3lc40802g |
| [16] | Dong G F, Xie SQ, Zhu XM, et al. Nutri-toxicological effects of cyanobacteria on fish[J]. Acta Ecologica Sinica, 2012, 32 (19): 6233–6241. DOI:10.5846/stxb |
| [17] | Wang Z, Yang J, Zhou J, et al. Composition and structure of bacterial communities in waste water of aquatic products processing factories[J]. Research Journal of Biotechnology, 2014, 9 (2): 65–70. |
| [18] | Swan BK, Chaffin MD, Martinez-Garcia M, et al. Genomic and metabolic diversity of Marine Group Ⅰ Thaumarchaeota in the mesopelagic of two subtropical gyres[J]. PLoS One, 2014, 9 (4): e95380. DOI:10.1371/journal.pone.0095380 |
| [19] | Finneran KT, Johnsen CV, Lovley DR. Rhodoferax ferrireducens sp. nov., a psychrotolerant, facultatively anaerobic bacterium that oxidizes acetate with the reduction of Fe(Ⅲ)[J]. International Journal of Systematic and Evolutionary Microbiology, 2003, 53 (3): 669–673. DOI:10.1099/ijs.0.02298-0 |
| [20] | Madigan MT, Jung DO, Woese CR, et al. Rhodoferax antarcticus sp. nov., a moderately psychrophilic purple nonsulfur bacterium isolated from an Antarctic microbial mat[J]. Archives of Microbiology, 2000, 173 (4): 269–277. DOI:10.1007/s002030000140 |
| [21] | Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome[J]. Science, 2016, 353 (6305): 1272–1277. DOI:10.1126/science.aaf4507 |
| [22] | Ma X, Zhao C, Gao Y, et al. Divergent taxonomic and functional responses of microbial communities to field simulation of aeolian soil erosion and deposition[J]. Molecular Ecology, 2017, 26 (16): 4186–4196. DOI:10.1111/mec.2017.26.issue-16 |
| [23] | Louca S, Jacques S, Pires APF, et al. Functional structure of the bromeliad tank microbiome is strongly shaped by local geochemical conditions[J]. Environ Microbiol, 2017, 19 (8): 3132–3151. DOI:10.1111/emi.2017.19.issue-8 |