




2. 中国科学院微生物研究所 植物基因组学国家重点实验室,北京 100101
2. State Key Laboratory of Plant Genomics,Institute of Microbiology,Chinese Academy of Sciences,Beijing 100101
稻瘟菌(Magnaporthe oryzae)能够在水稻生长的任何时期对水稻的不同部位进行侵染,造成水稻大量减产[1]。稻瘟菌所引起的稻瘟病,现已在全球范围蔓延,如中国、美国、韩国和日本等地,直接威胁世界粮食安全[1-5]。稻瘟菌全基因组测序的完成及序列公布[2-4],使得对稻瘟菌相关的研究更加深入,也更有利于植物和稻瘟菌互作的分子机制研究。在稻瘟菌对植物的侵染过程中,众多分泌蛋白参与其中,发挥着重要的作用。例如,GAS1和GAS2是稻瘟菌附着胞形成中特异性表达的两个基因,可以编码小型蛋白,并分泌到细胞质中[6]。GAS1或者GAS2基因的缺失,都会降低稻瘟菌附着胞的形成能力以及降低该菌的侵染能力[6]。胞外几丁质结合蛋白CBP1的研究表明,该蛋白含有两个几丁质结合结构域,在稻瘟菌感知外界环境及附着胞形成过程中具有重要的作用[7]。
分泌蛋白组(secretome)一词,在对枯草杆菌分泌蛋白的研究中首次提出[8]。分泌蛋白普遍存在于动植物中,功能多种多样,具有免疫、获取营养、重塑细胞壁环境及信号感知等功能[9]。通常情况下,含有信号肽并且通过内质网-高尔基体途径分泌的蛋白质集合,称之为分泌蛋白组[9, 10]。相应的蛋白称之为经典分泌蛋白(classical secreted protein)[9]。然而在高等生物中,如在拟南芥全基因组的研究中发现,预测18%的蛋白质为可分泌蛋白[10],但大多数预测的分泌蛋白质并不具备经典信号肽[10, 11]。这些不具备经典信号肽的可分泌蛋白被称为无信号肽分泌蛋白(leaderless secreted proteins)[11]。
随着多种真菌基因组测序的完成,越来越多的真菌分泌蛋白组以实验或者计算机预测的方式获得[12]。如黑曲霉(Aspergillus niger)[13]、白色念珠菌(Candida albicans)[14]、核盘菌(Sclerotinia sclerotiorum)[15]、玉米黑粉菌(Ustilago maydis)[16]、白腐菌(Phanerochaete chrysosporium)[17]以及禾谷镰刀菌(Fusarium graminearum)[18]等真菌的分泌蛋白组都已有报道。虽已有文章报道预测的稻瘟菌分泌蛋白组[19, 20],但是基本只是获得经典分泌蛋白的数目。已报道的稻瘟菌分泌蛋白组预测文章,未完成经典分泌蛋白大数据集的相关分析,如GO功能富集、KEGG代谢通路分析。另外,为了深入的研究稻瘟菌分泌蛋白与植物互作的关系,从稻瘟菌全基因组水平出发,获得可降解植物细胞壁等成分的经典分泌蛋白是十分必要的。值得一提的是,无信号肽分泌蛋白现在越来越被研究者所重视。但是关于稻瘟菌无信号肽分泌蛋白的预测,目前还没有相关的报道。
有鉴于此,本研究综合使用SignalP、TMHMM、big-PI Predictor、Protcomp和SecretomeP多种生物信息学分析软件,开发了新的分泌蛋白预测流程,对稻瘟菌经典分泌蛋白和无信号肽分泌蛋白进行了重新定义。同时,编写了Python脚本程序进行信息提取以及相关结构域数目的统计。在此基础上,进一步完成了对经典分泌蛋白的GO功能富集、KEGG通路以及结构域分析。同时,利用从稻瘟菌全基因组注释中所获取的众多信息,进行大规模的筛选,对有可能降解植物成分的稻瘟菌经典分泌蛋白进行预测,期望为进一步开展该菌分泌蛋白的功能研究以及研究水稻-稻瘟菌互作的分子机制提供有利的借鉴。
1 材料与方法 1.1 材料稻瘟菌的全蛋白组序列来源于Broad institute数据库(http://www.broadinstitute.org/annotation/genome/)。稻瘟菌本地blast比对数据库来自于KOBAS数据源(http://kobas.cbi.pku.edu.cn/site/download_fasta.jsp)。
本地化blast版本号为blast-2.2.30+,下载于NCBI(ftp://ftp.ncbi.nlm.nih.gov)。独立SignalP 4.1软件包下载自(http://www.cbs.dtu.dk/services/SignalP/)。独立TMHMM-2.0软件包下载自(http://www.cbs.dtu.dk/services/TMHMM/)。big-PI Predictor在线软件(http://mendel.imp.ac.at/sat/gpi/gpi_server.html)。
ProtComp 9.0在线软件(http://linux1.softberry.com/berry.phtml?topic=protcompan&group=programs&subgroup=proloc)。SecretomeP 2.0在线软件(http://www.cbs.dtu.dk/services/SecretomeP/)。
1.2 方法 1.2.1 分泌蛋白确定方法经典的分泌蛋白一般具有以下几个特征:具有信号肽、无跨膜结构域(N端40个氨基酸中含一个跨膜结构域除外)、无GPI锚定位点以及不含线粒体、叶绿体等细胞器的定位信号[11]。
本研究使用SignalP软件[21]、TMHMM软件[22]、big-PI Predictor软件[23]和Protcomp软件进行稻瘟菌经典分泌蛋白的预测。首先使用SignalP及TMHMM软件,对经典分泌蛋白信号肽和跨膜结构域进行大规模筛选。然后,使用big-PI Predictor软件[23]对具有信号肽且含有0或1个跨膜结构域的氨基酸序列进行GPI锚定位点预测。最后,使用Protcomp软件完成对蛋白的亚细胞定位预测。经过上述4步的严格筛选,得到蛋白基本可认为是经典分泌蛋白。
稻瘟菌无信号肽分泌蛋白的预测,使用最新的SecretomeP 2.0软件[24]进行筛选。筛选标准为:不具备信号肽,SecP值大于等于0.5。编写相应的Python脚本进行信息的提取和统计。
1.2.2 GO功能富集、KEGG通路分析及相关统计经典分泌蛋白的GO功能分析。首先,使用Python脚本语言提取经典分泌蛋白的氨基酸序列。然后,使用在线软件agriGO(http://bioinfo.cau.edu.cn/agriGO/analysis.php)对经典分泌蛋白进行GO功能富集分析,选取Fisher统计模型进行计算。
(2)对经典分泌蛋白进行KEGG代谢通路分析。首先构建本地化的blast平台,blast版本号为blast-2.2.30+。然后将稻瘟菌比对数据库格式化。使用blastp模式将分泌蛋白序列比对到稻瘟菌数据库中,E value设置为10-5。将blast比对结果导入到KOBAS软件(http://kobas.cbi.pku.edu.cn/home.do),进行KEGG差异通路分析。编写Python程序,提取相关信息并进行差异倍数计算。
稻瘟菌全基因组注释信息以及结构域信息来源于Broad institute数据库。编写Python脚本程序进行提取、相关统计以及大规模筛选,从而完成可降解植物细胞壁等成分的分泌蛋白预测。通过编写Python脚本,进行结构域的提取和统计。
1.2.3 原位(in planta)表达谱验证稻瘟菌分泌蛋白原位表达谱数据整合自Chen等[25]的研究。本实验通过提取原位分泌蛋白表达谱数据并与本研究所得假定分泌蛋白进行验证,进一步确定假定分泌蛋白的原位表达情况。
2 结果 2.1 稻瘟菌中经典分泌蛋白的预测稻瘟菌中共含有11 069条蛋白序列。首先使用本地化的SignalP 4.1软件对上述蛋白序列进行信号肽预测。SignalP软件可以对氨基酸序列的信号肽存在情况以及信号肽切割位点进行预测。为了保持软件预测敏感性的同时避免预测错误,软件D值采用系统默认。结果(图 1)表明,1 576条蛋白含有N端经典信号肽,约占全部序列的14.24%。

|
| 图 1 稻瘟菌分泌蛋白组预测分析流程 |
使用TMHMM-2.0软件对具有N端经典信号肽的蛋白序列进行跨结构域预测,结果(图 2)发现有1 278条蛋白序列不含有跨膜结构域,217条蛋白含有一个跨膜结构域,30条蛋白序列含有2个跨膜结构域,51条蛋白序列含有3个及3个以上的跨膜结构域,分别占蛋白总数(1 576条)的81%、14%、2%和3%。由于TMHMM 程序可能无法将信号肽序列和跨膜结构域区分开来[17, 18],因此将含有一个跨膜结构域及不含有跨膜结构域的序列初步认定为经典分泌蛋白,两部分蛋白共计1 495条。利用big-PI Predictor软件对上述1 495条蛋白序列进行GPI脂锚定蛋白预测,结果(图 1)显示,1 410个蛋白不具备GPI脂锚定位点,85个蛋白具有脂锚定位点。

|
| 图 2 1 576个含有信号肽的蛋白质跨膜结构域预测结果 |
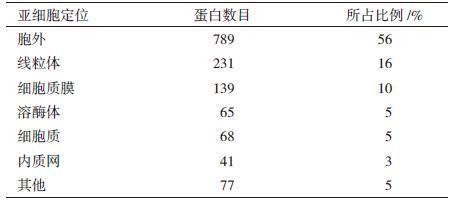
Protcomp软件对上述1 410个不具GPI脂锚定位点的蛋白质进行亚细胞定位预测。结果(表 1)表明,共有789个蛋白属于胞外分泌蛋白,其它621个蛋白并不分泌到胞外。621个不分泌到胞外的蛋白中,最多的是转运到线粒体(16%),其次是细胞质膜(10%)、溶酶体(5%)、细胞质(5%)和内质网(3%)。另外转运到高尔基体、过氧化物酶体、细胞核和液泡的蛋白占5%,共计77个。
通过上述的生物信息学流程分析,最终在稻瘟菌中获得了789个经典分泌蛋白(图 1)。通过对789个蛋白的氨基酸数目进行统计,结果(图 3)表明更多的经典分泌蛋白长度集中于100-500 aa,占经典分泌蛋白总量的79.84%。通过对数据的指数拟合(R2=0.6)显示,随着经典分泌蛋白长度的增加,其蛋白数目越来越少。由此可以推测,大多数的经典分泌蛋白质属于小型蛋白,所含氨基酸数目一般较少。

|
| 图 3 稻瘟菌789个经典分泌蛋白氨基酸长度分析 |
获取稻瘟菌789个经典分泌蛋白的基因号,使用AgriGO软件进行GO功能富集分析,设置稻瘟菌全基因组作为背景值,选取Fisher统计模型进行计算。通过对789个经典分泌蛋白的功能富集(图 4)发现,这些蛋白质多参与到分泌途径以及和宿主的相互作用中。由此可知,经典分泌蛋白在稻瘟菌和宿主互作过程中发挥着重要的作用。

|
| 图 4 GO富集差异生物学过程分析 |
为了完成经典分泌蛋白的KEGG通路分析。首先,使用本地化的blast将789个经典分泌蛋白序列比对到稻瘟菌数据库中。然后,使用在线软件KOBAS对数据进行分析,通过一系列复杂计算最终得到差异KEGG通路。结果(图 5)表明糖类代谢相关途径发生了显著的变化。如淀粉和蔗糖代谢、半乳糖代谢、戊糖和糖醛酸转化途径以及其它多糖的降解。由于稻瘟菌侵染水稻过程中需要大量的能量,因此获取能量对于稻瘟菌的生存极为重要。由此可以推测,许多经典分泌蛋白有可能参与到糖代谢途径中,为稻瘟菌的生存及侵染提供足够的能量。除上述途径外,氰基氨基酸代谢途径(Cyanoamino acid metabolism)也发生了显著的变化。这些结果表明,稻瘟菌分泌蛋白参与糖代谢途径可能是其成功侵染寄主的一个重要机制。

|
| 图 5 经典分泌蛋白差异KEGG通路分析 *P-value≤0.05;**P-value≤0.01 |
编写Python程序,从稻瘟菌全基因组中提取789个经典分泌蛋白的基因组注释,并通过编写的程序筛选出相应的可降解植物细胞壁等成分的经典分泌蛋白。经过大规模的筛选,本研究发现了156个分泌蛋白有可能具有降解植物细胞壁等成分的功能(表 2)。
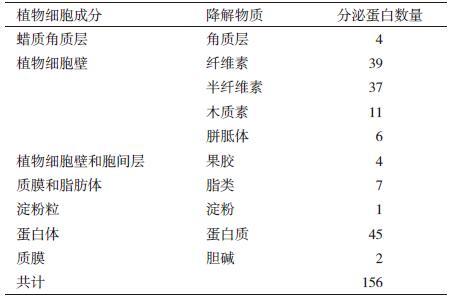
稻瘟菌分泌蛋白组中存在着许多降解植物蜡质层以及细胞壁结构的分泌蛋白和酶类。本研究发现,4个经典分泌蛋白能够特异性地降解植物的角质层。可降解植物纤维素的经典分泌蛋白数目较多,有39个。在降解植物纤维素的分泌蛋白中,有15个预测为内切糖苷酶,8个预测为可降解纤维二糖。半纤维素主要的两种成分为阿拉伯糖和木聚糖。在经典分泌蛋白中预测到37个可降解半纤维素的分泌蛋白。另外,还有11个预测为可降解木质素相关成分,这些分泌蛋白为漆酶、氧化物酶以及阿魏酸酯酶。4个经典分泌蛋白预测为果胶酯酶和果胶裂解酶,可降解植物细胞壁和胞间层之间的果胶。胼胝体存在于植物的胞间连丝以及韧皮部,是由β-1,3 葡聚糖形成的多聚物。在植物响应物理伤害以及病原菌侵染过程中起很重要的作用。本研究发现了6个经典分泌蛋白具有降解胼胝体的功能。
除降解植物蜡质层以及细胞壁的分泌蛋白外,本研究还发现了可降解植物蛋白质、脂类、淀粉以及胆碱的分泌蛋白(表 2)。通过对稻瘟菌全基因组注释信息的筛选,共有45个经典分泌蛋白可以降解蛋白质,该类分泌蛋白多为蛋白酶及肽酶。另外,7个经典分泌蛋白预测可以降解脂类物质。降解胆碱及淀粉的分泌蛋白数目较少,分别为2个和1个。
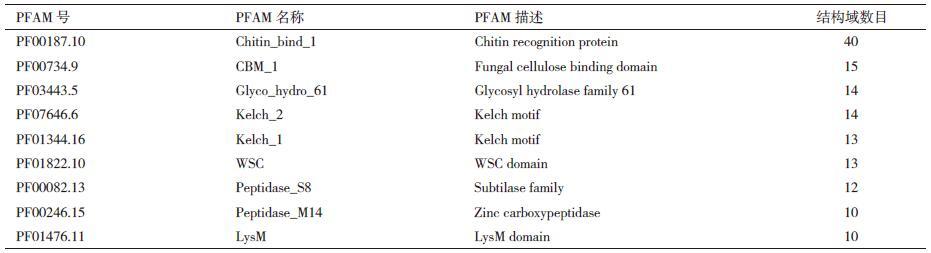
2.4 稻瘟菌经典分泌蛋白结构域分析从Broad institute数据库中提取稻瘟菌789个经典分泌蛋白的结构域注释,编写Python脚本语言,进行相关信息的统计。根据统计结果(表 3)可知,789个经典分泌蛋白中共有278个分泌蛋白存在功能结构域。其中,90个分泌蛋白具有两个及两个以上的结构域。另外,本研究统计了出现次数较多的结构域,见表 3。数目最多的结构域为Chitin_bind_1(PF00187.10),即几丁质结合结构域。其次为CBM_1(PF00734.9)、糖水解酶61家族(PF03443.5)以及Kelch相关结构域(PF07646.6,PF01344.16)。
另外,通过对789个经典分泌蛋白中所有结构域的筛选发现,含有的糖基水解酶家族相关结构域最多,约占结构域总数的20%(82个)。具体结构域分布见图 6。例如,PF04616结构域属于糖苷水解酶43家族,具有阿拉伯呋喃糖酶以及木糖酶活性。

|
| 图 6 稻瘟菌经典分泌蛋白中糖基水解酶家族结构域 |
以稻瘟菌全蛋白组氨基酸序列作为实验对象,使用SecretomeP软件进行分析,从而预测潜在的无信号肽分泌蛋白。本研究筛选结果表明,约有4 341个假定分泌蛋白满足上述条件,约占稻瘟菌总蛋白的39%。
为了增加实验的可信度,本实验从4 341个假定无信号肽分泌蛋白中选取了187个(SecP值≥0.9)进行了结构域分析(图 7)。选取的187个假定分泌蛋白中,只有57个具有结构域。57个具有结构域的假定无信号肽分泌蛋白中,含有最多的结构域为锌指结构域(PF00096.17)和氯霉素抗性引导肽结构域(PF08077.2)。在含有结构域的无信号肽分泌蛋白中,绝大多数只含有一个结构域,并且结构域多种多样。由此可见,非经典分泌蛋白应该具有更加多样化的功能。

|
| 图 7 非经典分泌蛋白(SecP值0.9)结构域 |
本研究整合了Chen等[25]稻瘟菌-水稻原位表达谱的数据,从中获得了831个原位表达的稻瘟菌分泌蛋白基因。通过统计(图 8)发现,本研究所获得的789个假定经典分泌蛋白(CSPs)中,共有354个在原位表达谱中被检测到,占所有假定分泌蛋白的45%。与经典分泌蛋白不同的是,在原位表达谱中仅检测到55个非经典分泌蛋白。

|
| 图 8 经典分泌蛋白及非经典分泌蛋白原位(in planta)表达情况 In planta:稻瘟菌原位表达谱数据中表达的831个分泌蛋白;CSPs:本实验预测得到的789个经典分泌蛋白;LSPs:本实验预测得到的4 341个非经典分泌蛋白 |
稻瘟菌全基因组序列的释放,为其分泌蛋白的研究奠定了重要的数据基础。众所周知,真菌分泌蛋白与致病关系密切。因此,在全基因组水平研究稻瘟菌的分泌蛋白特点,将有助于全面了解其致病因子的概况。
目前完成测序的稻瘟菌菌株70-15[2]、98-06[3]、Y34[4]及P131[4]中,其基因组序列大多是保守的。若以70-15菌株基因组作为参照,Y34、P131及98-06三种菌株基因组的保守率达到96%以上。菌株Y34、P131和70-15三者相较,其各自基因组中分别含有51个、136个和71个特有基因。在上述特有基因中,13%的基因预测可编码分泌蛋白[4]。在属于菌株Y34和P131的特有基因中,19%的基因预测为含有信号肽的分泌蛋白。另外,预测菌株98-06中含有1732个分泌蛋白,其中645个被认为是候选的效应蛋白[3]。由此可见,分泌蛋白在各个稻瘟菌生理小种中普遍存在。
本研究使用生物信息学的方法,最终在稻瘟菌中获得了789个经典分泌蛋白。通过对经典分泌蛋白氨基酸数目的统计发现,大多数的经典分泌蛋白长度集中于100-500 aa,属于小型蛋白。另外,通过GO功能富集分析发现,经典分泌蛋白在植物-稻瘟菌互作过程中起着非常重要的作用。由此可以推测,经典分泌蛋白多为小型蛋白,因其结构较为简单,可更加方便的在植物-稻瘟菌互作中发挥作用。在其它致病菌中,分泌蛋白也多为小型蛋白。如Cladosporium fulvum 中的Avr4、Avr4E和Avr9基因,Melampsora lini 中的AvrL567A、AvrL567B等,这些基因的产物均为经典分泌蛋白且氨基酸数目皆小于200,同时能使植物产生过敏反应[26]。另外,通过稻瘟菌-水稻互作的原位表达谱数据验证发现,354个预测所得经典分泌蛋白发生了差异表达。更加有力地说明了稻瘟菌在入侵水稻过程中,经典分泌蛋白发挥着极其重要的作用。
稻瘟菌在侵染过程中会形成附着胞,依靠附着胞产生的瞬间高压,从而达到刺穿植物细胞壁的目的[1, 5, 27, 28]。同时,稻瘟菌会分泌大量可降解细胞壁的酶类,从而加快菌丝侵染[29]。已有研究表明,病原菌侵染过程中所产生的分泌蛋白,可以降解植物成分。如稻瘟菌中的XYL-6基因,该基因产物即为分泌蛋白,且具有木聚糖酶活性,能够降解水稻细胞壁[30]。另外,在白粉菌(Erysiphe cichoracearum)侵染拟南芥的研究中发现,果胶裂解酶基因PMR6对于白粉菌的致病是必须的[31]。该基因产物为具有果胶裂解酶活性的分泌蛋白,可以完成对拟南芥细胞壁的降解[31]。本研究通过稻瘟菌全基因组注释,共预测出156个经典分泌蛋白可能具有降解植物成分的功能。其中,绝大部分分泌蛋白是降解植物蜡质层和细胞壁等相关成分的酶类。如降解蜡质层的角质酶,降解纤维素的葡聚糖酶以及降解半纤维素的木聚糖酶等。
作为兼性活体寄生菌[1, 5],稻瘟菌侵染植物过程中需要消耗大量的能量。植物作为稻瘟菌的直接碳源,如何利用植物成分为菌自身提供能量显得十分重要。虽然已有研究表明分泌蛋白具有降解植物成分的功能,但分泌蛋白对于稻瘟菌糖代谢途径的贡献却所知甚少。本研究通过对789个经典分泌蛋白KEGG代谢途径的分析发现,淀粉和蔗糖代谢(starch and sucrose metabolism)、半乳糖代谢(galactose metabolism)、戊糖和糖醛酸转化途径(pentose and glucuronate interconversions)以及其他多糖的降解(other glycan degradation)等途径发生了极其显著的变化(图 6)。同时,本研究还发现经典分泌蛋白中存在种类多样且数量较多的糖基水解酶结构域(图 7)。这意味着,众多的分泌蛋白有可能参与到糖代谢途径当中,为稻瘟菌提供所需的能量从而助力入侵过程。
经典分泌蛋白具有N端的信号肽,由信号肽引导核糖体到达内质网,从而完成多肽的合成[9, 32]。除了含有信号肽的经典分泌蛋白外,还存在一种不含有信号肽的分泌蛋白[11]。该类分泌蛋白在动植物中普遍存在。例如,人的内质网、高尔基体中即存在大量不具有信号肽的蛋白质[32, 33]。真菌中已有关于不含有信号肽分泌蛋白的研究。例如,酿酒酵母(Saccharomyces cerevisiae)中的a因子[34],以及灰盖鬼伞(Coprinus cinereus)通过非经典途径分泌的两个半乳凝素[35]。本研究通过对稻瘟菌分泌蛋白组的预测,发现了很多潜在的非经典分泌蛋白。同时,在稻瘟菌-水稻互作表达谱中也检测到该类分泌蛋白的表达。众多非经典分泌蛋白的存在是否具有重要的生物学意义,或者具有何种重要的生物学意义,随着稻瘟菌分泌蛋白研究的不断加深,相信非经典分泌蛋白的功能会得到很好的诠释。
4 结论本研究通过设计生物信息学分析流程,重新定义了稻瘟菌分泌蛋白组。其中,经典分泌蛋白789个,无信号肽分泌蛋白4 341个。通过对经典分泌蛋白的大规模数据分析发现,经典分泌蛋白在与宿主互作及糖代谢过程中起到重要的作用。另外,发现了156个经典分泌蛋白具有可降解植物细胞壁等成分的功能。经典分泌蛋白结构域的预测发现,存在最多的结构域为糖水解酶结构域。进而佐证了经典分泌蛋白在糖代谢中发挥着重要作用这一结论。
| [1] | Wilson RA, Talbot NJ. Under pressure:investigating the biology of plant infection by Magnaporthe oryzae. Nature Review Microbiology , 2009, 7 (3) : 185–195. DOI:10.1038/nrmicro2032 |
| [2] | Dean RA, Talbot NJ, Ebbole DJ, et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature , 2005, 434 (7036) : 980–986. DOI:10.1038/nature03449 |
| [3] | Dong Y, Li Y, Zhao M, et al. Global genome and transcriptome analyses of Magnaporthe oryzae epidemic isolate 98-06 uncover novel effectors and pathogenicity-related genes, revealing gene gain and lose dynamics in genome evolution. PLoS Pathogens , 2015, 11 (4) : 1–30. |
| [4] | Xue M, Yang J, Li Z, et al. Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genetics , 2012, 8 (8) : 1–12. |
| [5] | Ribot C, Hirsch J, Balzergue S, et al. Susceptibility of rice to the blast fungus, Magnaporthe grisea. Journal of Plant Physiology , 2008, 165 : 114–124. DOI:10.1016/j.jplph.2007.06.013 |
| [6] | Xue C, Park G, Choi W, et al. Two novel fungal virulence genes specifically expressed in appressoria of the rice blast fungus. The Plant Cell , 2002, 14 : 2107–2119. DOI:10.1105/tpc.003426 |
| [7] | Kamakura T, Yamaguchi S, Saitoh K, et al. A novel gene, CBP1, encoding a putative extracellular chitin-binding protein, may play an important role in the hydrophobic surface sensing of Magnaporthe grisea during appressorium differentiation. Molecular Plant-Microbe Interactions , 2002, 15 (5) : 437–444. DOI:10.1094/MPMI.2002.15.5.437 |
| [8] | Tjalsma H, Bolhuis A, Jongbloed JDH, et al. Signal peptide-dependent protein transport in Bacillus subtilis:a genome-based survey of the secretome. Microbiol Mol Biol Rev , 2000, 64 (3) : 515–547. DOI:10.1128/MMBR.64.3.515-547.2000 |
| [9] | Choi J, Park J, Kim D, et al. Fungal secretome database:integrated platform for annotation of fungal secretomes. BMC Genomics , 2010, 11 (105) : 1471–2164. |
| [10] | Alexandersson E, Ali A, Resj? S, et al. Plant secretome proteomics. Front Plant Science , 2013, 4 : 9. |
| [11] | Agrawal GK, Jwa NS, Lebrun MH, et al. Plant secretome:Unlocking secrets of the secreted proteins. Proteomics , 2010, 10 (4) : 799–824. DOI:10.1002/pmic.v10:4 |
| [12] | Bouws H, Wattenberg A, Zorn H. Fungal secretomes—nature’s toolbox for white biotechnology. Applied Microbiology and Biotechnology , 2008, 80 (3) : 381–388. DOI:10.1007/s00253-008-1572-5 |
| [13] | Tsang A, Butler G, Powlowski J, et al. Analytical and computational approaches to define the Aspergillus niger secretome. Fungal Genetics and Biology , 2009, 46 . |
| [14] | Lee SA, Wormsley S, Kamoun S, et al. An analysis of the Candida albicans genome database for soluble secreted proteins using computer-based prediction algotithms. Yeast , 2003, 20 (7) : 595–610. DOI:10.1002/(ISSN)1097-0061 |
| [15] | Yajima W, Kav NNV. The proteome of the phytopathogenic fungus Sclerotinia sclerotiorum. Proteomics , 2006, 6 (22) : 5995–6007. DOI:10.1002/(ISSN)1615-9861 |
| [16] | Mueller O, Kahmann R, Aguilar G, et al. The secretome of the maize pathogen Ustilago maydis. Fungal Genetics and Biology , 2008, 45 . |
| [17] | Wymelenberg AV, Sabat G, Martinez D, et al. The Phanerochaete chrysosporium secretome:Database predictions and initial mass spectrometry peptide identifications in cellulose-grown medium. Journal of Biotechnology , 2005, 118 : 17–34. DOI:10.1016/j.jbiotec.2005.03.010 |
| [18] | Paper JM, Scott-Craig JS, Adhikari ND, et al. Comparative proteo-mics of extracellular proteins in vitro and in planta from the patho-genic fungus Fusarium graminearum. Proteomics , 2007, 7 (17) : 3171–3183. DOI:10.1002/(ISSN)1615-9861 |
| [19] | 陈继圣, 郑士琴, 郑武, 等. 全基因组预测稻瘟菌的分泌蛋白. 中国农业科学 , 2006, 39 (12) : 2474–2482. |
| [20] | 苏源, 李成云, 赵之伟, 等. 稻瘟菌基因组规模分泌蛋白的预测分析. 云南农业大学学报 , 2006, 21 (3) : 271–275. |
| [21] | Petersen TN, Brunak S, Heijne G, et al. SignalP 4. 0:discriminating signal peptides from transmembrane regions. Nature Methods , 2011, 8 (10) : 785–786. DOI:10.1038/nmeth.1701 |
| [22] | Krogh A, Larsson Bè, Heijne G, et al. Predicting transmembrane protein topology with a hidden markov model:application to complete genomes. Journal of Molecular Biology , 2001, 305 (3) : 567–580. DOI:10.1006/jmbi.2000.4315 |
| [23] | Eisenhaber B, Schneider G, Wildpaner M, et al. A Sensitive predictor for potential GPI lipid modification sites in fungal protein sequences and its application to genome-wide studies for Aspergillus nidulans, Candida albicans Neurospora crassa, Saccharomyces cerevisiae and Schizosaccharomyces pombe. Journal of Molecular Biology , 2004, 337 (2) : 243–253. DOI:10.1016/j.jmb.2004.01.025 |
| [24] | Bendtsen JD, Jensen LJ, Blom N, et al. Feature-based prediction of non-classical and leaderless protein secretion. Protein Engineering, Design and Selection , 2004, 17 (4) : 349–356. DOI:10.1093/protein/gzh037 |
| [25] | Chen SB, Songkumarn P, Venu RC. Identification and characteriza-tion of in planta-expressed secreted effector proteins from Magnap-orthe oryzae that induce cell death in rice. The American Phytopathological Society , 2013, 26 (2) : 191–202. |
| [26] | Rep M. Small proteins of plant-pathogenic fungi secreted during host colonization. FEMS Microbiology Letters , 2005, 253 (1) : 19–27. DOI:10.1016/j.femsle.2005.09.014 |
| [27] | Ebbole DJ. Magnaporthe as a model for understanding host-patho-gen interactions. Annual Review of Phytopathology , 2007, 45 : 437–456. DOI:10.1146/annurev.phyto.45.062806.094346 |
| [28] | Giraldo MC, Dagdas YF, Gupta YK, et al. Two distinct secretion systems facilitate tissue invasion by the rice blast fungus Magnap-orthe oryzae. Nature Communications , 2013, 4 : 1996. |
| [29] | Mendgen K, Hahn M, Deising H. Morphogenesis and mechanisms of penetration by plant pathogenic fungi. Annual Review of Phytopathology , 1996, 34 : 367–386. DOI:10.1146/annurev.phyto.34.1.367 |
| [30] | Vogel JP, Raab TK, Schiff C, et al. PMR6, a pectate lyase-like gene required for Powdery mildew susceptibility in Arabidopsis. The Plant Cell , 2002, 14 (9) : 2095–2106. DOI:10.1105/tpc.003509 |
| [31] | Wu SC, Halley JE, Luttig C, et al. Identification of an endo--1, 4-D-xylanase from Magnaporthe grisea by gene knockout analysis, purification, and heterologous expression. Applied and Environmental Microbiology , 2006, 72 (2) : 986–993. DOI:10.1128/AEM.72.2.986-993.2006 |
| [32] | Lum G, Min XJ. FunSecKB:the fungal secretome knowledge base. Database , 2011, 2011 : bar001. |
| [33] | Scott M, Lu G, Hallett M, et al. The Hera database and its use in the characterization of endoplasmic reticulum proteins. Bioinformatics , 2004, 20 (6) : 937–944. DOI:10.1093/bioinformatics/bth010 |
| [34] | Chen P, Sapperstein SK, Choi JD, et al. Biogenesis of the Saccharomyces cerevisiae mating pheromone a-factor. The Journal of Cell Biology , 1997, 136 (2) : 251–269. DOI:10.1083/jcb.136.2.251 |
| [35] | Boulianne RP, Liu Y, Aebi M, et al. Fruiting body development in Coprinus cinereus:regulated expression of two galectins secreted by a non-classical pathway. Microbiology , 2000, 146 (8) : 1841–1853. DOI:10.1099/00221287-146-8-1841 |