




2. 深圳市坪山新区环境监测站,深圳 518118
2. Pingshan Environmental Monitoring Station,Shenzhen 518118
苦荞(tartary buckwheat)是一种蓼科荞麦属双子叶植物,又名鞑靼荞麦[1](Fagopyrum tataricum),是药食两用的粮食珍品,原产于我国西南部的四川凉山地区,目前在西北和西南等地区广有种植[2]。苦荞不仅营养价值丰富,还含有黄酮类等活性成分,具有降糖脂、降胆固醇、抗氧化、清除自由基和消炎等功效[3]。研究表明,苦荞在萌发后氨基酸更为均衡,萌发过程可以富集γ-氨基丁酸(GABA)、黄酮和芦丁[4]。目前,国内外在苦荞芦丁和蛋白分离及功能性方面已有较多的研究[5-7],但发芽苦荞的分子生物学研究较少,造成其分子标记开发、遗传图谱构建、生长发育及其抗逆机理方面的研究相对滞后。在特定基因方面,赵海霞等[8]采用半定量RT-PCR分析发芽6 d苦荞其黄酮合成途径中主要关键酶基因,以及其转录因子基因相对表达水平;李成磊等[9]用同源克隆和cDNA末端快速克隆技术,获得苦荞CYP81家族同源基因FtP450-R4。在物种多样性方面,高帆等[10]用正交设计法筛选适用于苦荞SSR标记分析的PCR反应体系,筛选出19对引物进行苦荞遗传多样性分析。
近年来,包括基因组、转录组、蛋白质组等各种组学技术在揭示细胞生理活动规律和生物代谢机理的研究中起着越来越重要的作用,而转录组学是率先发展起来及应用最为广泛的技术,能全面快速地获得某一物种特定细胞或组织在某一状态下的基因表达情况[11]。同时,随着高通量测序技术的发展,测序成本的降低,基于高通量测序技术的转录组分析逐渐成为非模式植物中发掘功能基因的一种有效手段[12]。因此,本研究以Illumina SolexaHiseq 2500高通量测序技术对发芽苦荞进行转录组测序,旨在获得更多发芽苦荞的转录本和更为全面的转录组信息,发掘苦荞发芽过程中的重要基因表达。
1 材料与方法 1.1 材料实验材料为苦荞发芽子叶,样品由安徽科技学院食品药品学院食品科学与工程课题组提供。将苦荞种子(内蒙古自治区乌兰察布市生产)以去离子水清洗后,用1%的次氯酸钠消毒15 min后冲洗至pH中性,于去离子水中30℃浸泡2 h,置于铺有两层滤纸的培养皿中,每8 h喷去离子水1次,在30℃的培养箱内避光发芽2 d后,选取长势良好、健康的植株子叶,迅速将其放入纸带内,立即经液氮速冻后保存于实验室超低温冰箱中备用。提取嫩叶的RNA作为本次实验的需要的RNA。
1.2 方法 1.2.1 RNA提取样品在液氮中研磨至粉末状,加入TrizoI试剂混合均匀,利用TrizoI法提取试验材料苦荞发芽叶片的总RNA。cDNA文库的构建参考文献[13]的方法。
1.2.2 文库的建立与测序提取样品总RNA后,用带有Oligo(dT)的磁珠富集真核生物mRNA。加入fragmentaion buffer将 mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成第一条cDNA链,然后加入缓冲液、dNTPs、RNase H 和DNA polymerase I合成第二条cDNA链,在经过QiaQuick PCR试剂盒纯化并加EB缓冲液洗脱之后做末端修复、加polyA并连接测序接头,然后进行琼脂糖凝胶电泳并分离纯化,最后进行PCR扩增,得到建好的测序文库并将其用Illumina HiSeq 2500进行双端测序(paired-end)。
1.2.3 数据的过滤因为测序得到的reads(即Raw reads)并不都是有效的,里面含有带接头或污染的reads,这些reads会影响组装和后续分析,我们必须对下机的reads进行过滤,得到有效reads(即Clean reads)。
1.2.4 组装最后利用Trinity软件对Clean reads进行拼接。通过reads overlap关系得到的不含N的组装片段Contig,然后以paired-end reads将来自同一转录本的不同Contig连接,得到两端不能再延长的非冗余序列(即unigenes)。
1.2.5 功能注释首先通过blastn程序将unigenes比对到NCBI-Nt核酸数据库。通过blastx程序将unigenes比对到蛋白质数据库。蛋白质数据库包括NR、Swiss-Prot、KEGG、GO和KOG,E值<1e-5。其 中,unigenes通过COG、GO和KEGG数据库的分类的参考文献[14]的方法。
1.2.6 CDS预测通过BLAST软件将unigenes序列与蛋白质数据库比对,得到unigenes编码区的核酸序列(序列方向5′-3′)和氨基酸序列。后以orfpredict软件预测没有比对到蛋白质数据库的unigenes的CDS序列和氨基酸序列。
1.2.7 SSR位点的筛选SSR位点的筛选利用MISA软件在所有unigenes中搜索SSR位点,参数设置如下:单核苷酸、二核苷酸至少重复次数为10,三核苷酸、四核苷酸、五核苷酸和六核苷酸至少重复次数均为5,对查找的SSR类型进行特征分析。
2 结果 2.1 荞麦转录组数据的组装采用Illumina HiSeq 2500高通量测序技术对荞麦发芽嫩叶组织转录组进行测序,共得到42 953 962条长度为125 bp的Raw reads。去除adapter和低质量reads后,得到42 818 102条Clean reads。因为Clean reads中Q20的百分率为93.8%(>90%),所以质量合格,可进行后续分析。对Clean reads序列进行组装,采用Trinity软件,在拼接序列去重复后共获得71 366条长度大于200 bp的contig,长度79 Mb。最大长度、平均长度及N50分别为15 658、1 102和1 748 bp。取每条Loci下最长的转录本作为unigenes,得到了45 278条unigenes,总长度为39 Mb,平均长度与N50分别为862 bp和1 476 bp。其中,大于2 000 bp的序列共有4 426条( 图 1 ),占unigenes总数的9.78%,说明测序质量较好。
另外,GC含量是基因组碱基序列的重要特征之一,能反映基因的结构、功能和进化信息,GC分布不均匀导致基因组不同GC含量序列其性质和功能也有差异。荞麦发芽嫩叶组织的GC含量平均值为42.50%,其中GC含量过高(大于80%)或过低(小于20%)的unigenes不存在,GC含量基本呈正态分布( 图 2 ),从另一方面说明测序质量较好。

|
| 图 1 Unigenes长度分布图 |

|
| 图 2 Unigenes GC含量分布图 |
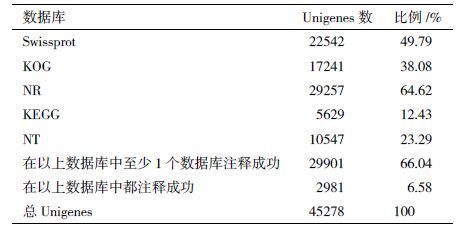
使用BLAST程序将组装得到的unigenes与NT、NR、KOG、Swissprot、KEGG数据库进行比对,进行unigenes的序列相似性分析。结果( 表 1 )显示,在NR注释成功的unigenes的数量最多(64.62%),其后依次是Swissprot(49.79%),KOG(38.08%),KEGG(12.43%)。对该4组数据库进行拓扑分析,结果( 图 3 )表明,共有2 981条unigenes四条数据库中同时标注成功,占总unigenes数的6.58%。并且在以上4条数据库中至少1条数据库注释成功的unigenes有29 901条,占总unigenes数的66.04%。其中,Swissprot数据库有少部分(48条)超出NR数据库范围,这可能是由于注册过程中两种数据库对于特定基因的更新不同步所致。以NR数据库为例进行分析,结果( 图 4 )表明,12 779条unigenes在NR数据库中可找到相似序列。在大于4%相似序列匹配的近缘物种中,葡萄(Vitis vinifera)所占比例最高(23.87%),其后依次是可可(Theobroma cacao,11.58%),杨毛 果(Populus trichocarpa,6.88%),桃(Prunus persica,6.82%)及番茄(Solanum lycopersicum,4.54%),其他物种占17.26%。

|
| 图 3 注释上各数据库韦恩图 |

|
| 图 4 NR注释物种分布图 |
真核生物蛋白相邻类的聚簇(clusters of orthologous groups for eukaryotic complete genomes,KOG)是对基因产物进行直系同源分类的数据库[15],将发芽苦荞与KOG数据库进行对比,可预测unigenes功能并进行分类统计。结果表明,共有17 241条unigenes(占unigenes总数的38%)被注释到24种KOG分类中 ( 图 5 中用A-Z表示)。从图中可以看出unigenes涉及的KOG功能类别比较全面,涉及了大多数的生命活动。其中,“一般功能基因”是最大类别,包含2 197条unigenes,占被注释到unigenes总数的12.74%;其次是“信号传导机制”,包含2 059条unigenes;而“未命名蛋白”(2个)和“核结构”(12个)类基因较少;其他类别的基因表达丰度都各不相同。

|
| 图 5 KOG分类图 A:RNA加工与修饰 ;B:染色质结构与变化 ;C:能量产生与转化;D:细胞周期调控与分裂,染色体重排 ;E:氨基酸运输与代谢 ;F:核苷酸运输与代谢;G:碳水化合物运输与代谢;H:辅酶运输与代谢;I:脂类运输与代谢;J:翻译,核糖体结构与生物合成;K:转录;L:复制、重组与修复;M:胞壁/膜生物发生;N:细胞运动;O:蛋白质翻译后修饰与转运,分子伴侣;P:无机离子运输与代谢;Q:次生产物合成,运输及代谢;R:一般功能基因;S:功能未知;T:信号传导机制;U:胞内分泌与膜泡运输;V:防御机制;W:胞外结构;X:未命名蛋白;Y:核结构;Z:细胞构架 |
基因本体论(gene ontology,GO)是一个国际标准化的基因功能分类数据库,用于全面地描述不同生物中基因的生物学特征[16]。结合GO数据库对发芽苦荞的unigenes进行功能分类,可从宏观上认识发芽苦荞表达基因的功能分布特征。结果( 图 6 )表明,有22 376条unigenes被注释上GO分类,其中,样本基因数量在10 000条以上且功能在参与的生物学过程(biological process)分类中主要聚集于细胞进程(cellular process)(14 033个)和代谢过程(metabolic process)(12 612个);在细胞组分(cellular Component)主要聚集于细胞(cell)(16 492个)和细胞成分(cell part)(16 492个);在分子功能(molecular function)分类中主要聚集于蛋白结合(binding)(12 890个)和催化活性(catalytic activity)(11 714个)。

|
| 图 6 GO注释上的基因分布 |
(kyoto encyclopedia of genes and genomes,KEGG)是系统分析基因产物在细胞中的代谢途径以及基因产物功能的数据库。根据KEGG数据库的注释信息能进一步得到unigenes的Pathway注释[17]。结合KEGG数据库,对发芽苦荞的unigenes可能参与或涉及的代谢途径进行了统计分析。结果表明,3 662条unigenes参与到328个代谢通路中,其中包含unigenes最多的代谢通路是核糖体(ko03010)( 表 2 ),共有410条unigenes,这可能是因为苦荞萌发时,预存在种子里的mRNA指导合成部分蛋白质,形成各种酶,接着这些酶进一步促使新的mRNA的生成,合成更多的蛋白质,导致核糖体以及线粒体也同时形成[18];其次是碳水化合物代谢(ko01200),包含176条unigenes。而参与氧化磷酸化(ko00190)的代谢通路的unigenes共有143条。

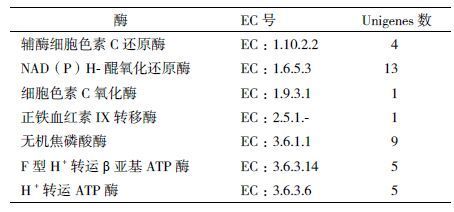
苦荞发芽过程可以富集γ-氨基丁酸(GABA),而GABA代谢过程与氧化磷酸化过程密不可分,当植物线粒体氧化磷酸化作用减弱,还原电位增加时,琥珀酸半醛脱氢酶活性降低。从而消弱了琥珀酸半醛生成琥珀酸的反应,有利于发芽苦荞中GABA的合成积累[19]。因此,结合KEGG数据库,对pathway中关于氧化磷酸化通路中发掘到的unigenes进行注释,共统计筛选出38条参与氧化磷酸化合成的unigenes( 表 2 ),编码7个关键酶,其中4个unigenes编码辅酶细胞色素C还原酶;13个unigenes编码NAD(P)H-醌氧化还原酶;1个unigenes编码细胞色素C氧化酶;1个unigenes编码正铁血红素IX转移酶;9个unigenes编码无机焦磷酸酶;5个unigenes编码F型H+转运β亚基ATP酶;5个unigenes编码H+转运ATP酶。由氧化磷酸化代谢通路(ko00190)的注释结果( 图 7 )可以看出,除编号为1.6.99.3、2.7.4.1和3.6.3.10的基因外,其余基因均被注释成功。

|
| 图 7 氧化磷酸化代谢通路(ko00190)的注释结果 绿色方框内的数字表示被注释成功的基因 |
编码序列(coding sequence,CD- S)指完整的编码蛋白质序列,CDS的预测可对后续苦荞麦的基因功能研究和基因组图谱的绘制提供重要的资源。结果表明,通过与NR数据库的比对,得到的CDS序列44 995个,对未与NR数据库比对上的unigens,用orfpredict软件进行CDS的预测。CDS的长度分布如 图 8 所示。

|
| 图 8 CDS长度分布图 |
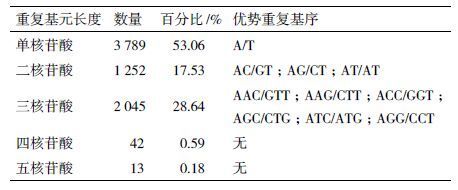
简单重复序列(simple sequence repeats,SSR)又称短串联重复序列,广泛存在于真核生物基因组中,一般采用SSR分子标记法对物种种质资源进行遗传多样性分析[20]。本实验利用MISA软件在发芽苦荞的71 366条unigenes中共搜索到7 141个SSR位点,占unigenes总序列的10.00%。SSR的类型丰富,单核苷酸至五核苷酸重复类型均存在,所占比例变化较大( 表 4 )。其中,单核苷酸重复所占比例最高,达到了53.06%;比例最低的是五核苷酸重复,仅为0.18%;二核苷酸重复和三核苷酸重复所占比例大致相当,分别为17.53%和28.46%。在检测到的SSR中,出现频率最高的10类基序为:A/T(3 744个)、AAG/CTT(690个)、AT/AT(607个)、AG/CT(536个)、ATC/ATG(357个)、ACC/GGT(271个)、AGG/CCT(176个)、AAC/GTT(166个)、AGC/CTG(154个)、AC/GT (107个)。上述SSR特征分析,有助于开展苦荞嫩叶组织及其同属物种的基因组差异分析、通用性标记开发和遗传图谱构建的研究。
本研究首次采用相对于454测序技术和SOLiD测序技术在测序成本和数据量输出方面更具优势的Illumina SolexaHiseq 2500高通量转录组测序平台[21],对发芽苦荞的转录组进行测序和功能分析。结果表明,经过预处理,各样本数据留存率均在99%以上,并且样本原始数据量均达到5 Gb以上序列平均长度约为117.91 bp,满足分析需求。并且序列组装后得到了45 278个unigenes,平均长度为862 bp,N50值(指从组装最长的unigenes依次向下求长度的总加和,当累加长度达到组装长度的一半时,对应的unigenes长度是N50长度)为1 746 bp,组装得到的长片段数量较多,组装效果较好[22]。此次序列组装的质量和长度可以满足转录组分析的基本要求。
45278个unigenes只有26 248个在Blast、同源性搜索中得到注释,剩下19 030个unigenes可能是由于较短而未与公共数据库中的序列比对上,也可能是非编码序列或者是新的基因[23]。利用KOG数据库对发芽苦荞unigenes进行基因功能分类,可从基因组水平上找寻直系同源体,预测未知ORF的生物学功能,可以大大提高基因功能注释的准确性。根据KEGG数据库对上述unigenes进行代谢途径分析,涉及328个具体的代谢途径分支,参与到发芽苦荞体内的核糖体代谢、碳水化合物代谢、氧化磷酸化等过程中,为进一步大量挖掘苦荞发芽过程中的重要表达基因,开展发芽苦荞的基因克隆及功能验证等研究提供了基础数据。其中GABA代谢过程与氧化磷酸化过程密不可分。
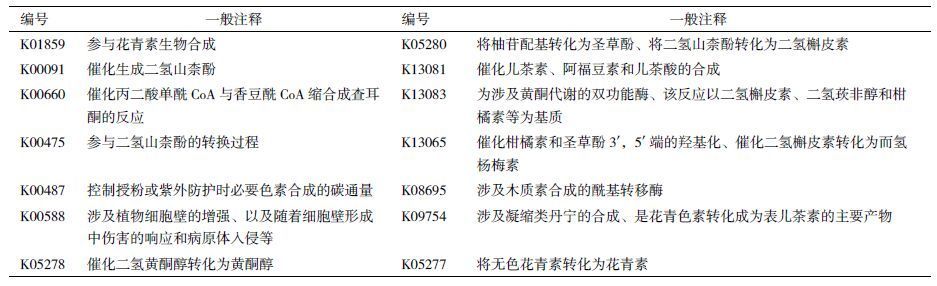
苦荞中含有黄酮类物质,其主要成分为芦丁。黄酮类化合物代谢途径中的相关基因,如 表 5 所 示[24, 25],但测序结果均未涉及,这与之前文献[4]中的结论并不一致,这可能与萌发阶段有关[26]。
本研究在发芽苦荞中发掘到7 141个SSR位点,其中单核甘酸和二核甘酸的重复占总数的70.59%,为保证SSR位点的潜在多态性,在筛选过程中对于三、四和五核甘酸的最小重复次数同样设置为5,一定程度上影响了这3类核昔酸重复在总SSR位点中所占比例。本研究结果为今后研究荞麦在发芽过程中相关基因的调控作用,特别是发芽过程中GABA代谢产物的代谢途径奠定了基础。
4 结论本研究通过Illumina SolexaHiseq 2500高通量测序,获得5.37 Gb的发芽苦荞转录组序列,拼接获得45 278条unigenes,发掘出38条参与氧化磷酸化的unigenes以及7 141个SSR位点。
| [1] | 顾娟. 荞麦淀粉理化特性及消化性研究[D]. 无锡:江南大学食品学院,2010. |
| [2] | 郭刚军, 何美节, 邹建云, 等. 苦荞黄酮的提取分离及抗氧化活性研究[J]. 食品科学, 2008, 29(12) : 373–376. |
| [3] | 张瑞. 苦荞黄酮及其降血糖活性研究[D]. 北京:中国农业科学院,2008. |
| [4] | 蔡马. 萌发对荞麦营养成分的影响研究[J]. 西北农业学报, 2004, 13(3) : 18–21. |
| [5] | 朱琳, 任清, 徐笑颖. 高速逆流色谱分离纯化苦荞中芦丁、槲皮素[J]. 食品科学, 2014, 35(3) : 47–50. |
| [6] | Kim SL, Park CH. Introduction and nutritional evaluation of buckwheat sprouts as a new vegetable[J]. Food Research International, 2004, 37(4) : 319–327. |
| [7] | Gao XN, Yao HY. Fractionation and characterization of tartary buckwheat flour proteins[J]. Food Chemistry, 2006, 1 : 90–94. |
| [8] | 赵海霞, 吴小峰, 白悦辰, 等. 苦荞芽期黄酮合成关键酶和MYB转录因子基因的表达分析[J]. 农业生物技术学报, 2012, 20(2) : 121–128. |
| [9] | 李成磊, 赵海霞, 温国琴, 等. 苦荞细胞色素CYP81家族同源基因FtP450-R4的克隆、分子鉴定及其功能分析[J]. 农业生物技术学报, 2015, 23(2) : 181–192. |
| [10] | 高帆, 等. 中国苦荞SSR分子标记体系构建及其在遗传多样性分析中的应用[J]. 中国农业科学, 2015, 6 : 1042–1053. |
| [11] | Maher CA, et al. Transcriptome sequencing to detect gene fusions in cancer[J]. Nature, 2009, 458(7) : 97–101. |
| [12] | Shu S, Chen B, Zhao X, et al. De novo sequencing and transcriptome analysis of Wolfiporiacocos to reveal genes related to biosynthesis of triterpenoids[J]. PLoS One, 2013, 8(8) : e71350. |
| [13] | Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis[J]. Nature Protocols, 2013, 8(8) : 1494–1512. |
| [14] | Guttikonda SK, et al. Whole genome co-expression analysis of soybean cytochrome P450 genes identifies nodulation-specific P450 monoox-ygenases[J]. BMC Plant Biology, 2010, 1 : 243. |
| [15] | Zhou Y, Gao F, Liu R, et al. De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus[J]. BMC Genomics, 2012, 13 : 266. |
| [16] | 杨楠, 等. 蜡梅花转录组数据分析及次生代谢产物合成途径研究[J]. 北京林业大学学报, 2012, 34(1) : 104–107. |
| [17] | 王晓锋, 何卫龙, 蔡卫佳, 等. 马尾松转录组测序和分析[J]. 分子植物育种, 2013, 11(3) : 385–392. |
| [18] | 谭保才, 等. 激动素对绿豆子叶多聚核糖体形成的促进作用及其与RNA合成的关系[J]. 植物学报, 1992, 9(10) : 74–76. |
| [19] | Shelp BJ, et al. Metabolism and functions of gamma-aminobutyric acid[J]. Trends Plant Sci, 1999, 4(7) : 446–452. |
| [20] | 刘峰, 王运生, 田雪亮, 等. 辣椒转录组SSR挖掘及其多态性分析[J]. 园艺学报, 2012, 39(1) : 168–174. |
| [21] | Kim SJ, Maeda T, Sarker MZ, et al. Identification of anthocyanins in the sprouts of buckwheat[J]. Journal of Agricultural and Food Chemistry, 2007, 55(15) : 6314–6318. |
| [22] | Xu Y, et al. Transcriptome and comparative gene expression analysis of Sogatella furcifera(Horváth)in response to southern rice black-streaked dwarf virus[J]. PLoS One, 2012, 7(4) : e36238. |
| [23] | Konishi T, et al. A linkage map of common buckwheat based on microsatellite and AFLP markers[J]. Fagopyrum, 2006, 2 : 1–6. |
| [24] | Kalra S, Puniya BL, Kulshreshtha D, et al. De novo transcriptome sequencing reveals important molecular networks and meta-holic pathways of the plant,Chlorophytum horivilianum[J]. PLoS One, 2013, 8(12) : e83336. |
| [25] | Schijlen EC, et al. Modification of flavonoid biosynthesis in crop plants[J]. Phytochemistry, 2004, 65(19) : 2631–2648. |
| [26] | Niu SH, Li ZX, Yuan HW, et al. Transcriptome characterization of Pinus tabuliformis and evolution of genes in the Pinus phylogeny[J]. BMC Genomics, 2013, 14 : 263. |